Le site des ophtalmologistes de France
Espace Encyclopédie
Encyclopédie de la vue

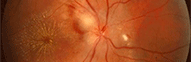




Maladie de Tay-Sachs
Maladie de Tay-Sachs
Gangliosidose GM2
Déficit en Hexosaminidase A
GM2-Gangliosidosis
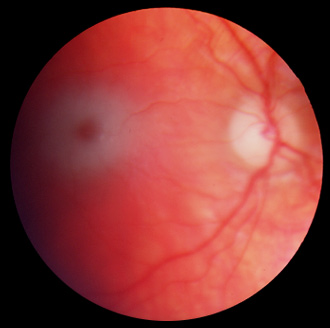
Maladie de Tay-Sachs
Cliché Pr Mathis CHU Rangueil Toulouse France
Définition
Cette maladie génétique rare fait partie du groupe des dyslipoïdoses, et aboutit à la cécité et le plus souvent à la mort dans l'enfance. Elle correspond à une thésaurismose de lipides (une accumulation), principalement dans le système nerveux de l'organisme, ce qui va entraîner des tableaux neurologiques sévères.
Elle fut décrite la première fois par Tay en 1881 puis par Sachs en 1887 qui la nommèrent "idiotie amaurotique familiale". Ces lipides sont des gangliosides GM2 qui s'accumulent par déficit d'une enzyme, la ß-N-acétylhexosaminidase A. Il existe des variantes de ce déficit.
Actuellement, à la suite de dépistages précoces, on assiste à une diminution des cette maladie dans certaines communautés (voir ci-dessous Dor Yeshorim).
Les dyslipoïdoses
Le groupe des dyslipoïdoses correspond à des phénomènes d'accumulation de sphingolipides dans les lysosomes cellulaires. Ce sont des sphingolipidoses lysosomiales.
Dans les mucopolysaccharidoses, il y a accumulation d'un sucre:
- syndrome de Hurler
- syndrome de Hunter
- syndrome de Scheie
- syndrome de Sanfilippo
- syndrome de Morquio
- syndrome de Maroteaux-Lamy
- syndrome de Sly
Les formes mixtes (lipide/sucre) sont les mucolipidoses (sialidoses).
Les sphingolipidoses donnent une accumulation de lipides dans les cellules ganglionnaires de la rétine et le tissu nerveux (neurolipidoses). En fonction du déficit biochimique, on observera des tableaux cliniques variés, tant dans l'âge d'apparition que dans la sévérité des signes ou le pronostic.
On décrit 4 types de dyslipoïdoses:
1) les gangliosidoses par accumulation de ganglioside:
- maladie de Tay-Sachs (déficit en ß-hexosaminidase A)
- maladie de Norman-Landing (déficit en gangliosido-ß-galactosidase)
2) les sphingomyélinoses par accumulation de sphingomyéline:
- maladie de Niemann-Pick (déficit en shpingomyélinase)
3) les cérébrosidoses par accumulation de cérébroside
- maladie de Sandhoff (déficit en ß-Hexosaminidase A.B)
- maladie de Fabry (déficit en alpha galactosidase A)
- cytosidose (déficit en lactosyl céramide ß-galactosidase)
- maladie de Gaucher (déficit en cérébroside ß-glucosidase)
- maladie de Durand ou fucocérébrosidose (déficit en alpha fucosidase)
- maladie de Farber (déficit en céramidase)
4) les sphingolipidoses sulfatiques par accumulation de sphingolipide sulfatique:
- maladie de Scholz (déficit en aryl-sulfatase A)
- maladie de Krabbe (déficit en galactosyl céramide ß-galactosidase)
Génétique
La maladie de Tay-Sachs a un transmission sous le mode autosomal récessif, et se retrouve dans certaines communautés: les Juifs Ashkénazes, les Canadiens français, les Cajuns de Louisiane. On retrouve dans ces populations un porteur du gène pour 27 individus, alors que dans les populations autres, la fréquence est de un pour 250. Il y a donc un risque multiplié par 10 de voir apparaître la maladie.
Le gène muté se trouve en 5q31.3-q33.1.
Clinique
On décrit chez les jeunes enfants de quelques mois, un sursaut inépuisable au bruit, avec un retard psycho-moteur et une hypotonie. L'examen de la vision des enfants est difficile à cause du syndrome neurologique, mais elle peut être assez bonne au début de l'évolution, pour rapidement s'aggraver et aboutir à la cécité. Celle-ci s'accompagne de nystagmus et de troubles oculo-moteurs. Le décès suivra, souvent avant l'âge de deux ans.
Il existe des formes moins graves qu'on découvre chez l'enfant de 5 ou 6 ans, avec troubles du comportement et ataxie locomotrice. Les formes de l'adulte peuvent être confondues avec une ataxie de Friedreich.
Le signe ophtalmologique princeps est la "tâche rouge cerise maculaire" bilatérale (cherry-red spot), décrite par Tay. La fovéola apparaît en effet rouge au sein d'une zone blanc-grisâtre: cet aspect est dû à la surcharge des cellules ganglionnaires par le ganglioside. Cette tâche apparaît entre la 4ème et la 12ème semaine après la naissance. Sa présence est d'autant plus fréquente que l'apparition de l'affection est précoce et que son évolution est plus grave.
En fait la fovéola rouge est normale mais, comme elle ne contient pas de cellules ganglionnaires, elle fait contraste avec la rétine pathologique gris-blanchâtre située autour d'elle.
On retrouve souvent ce signe dans les autres dyslipoïdoses.
On a constaté qu'une atrophie optique survenait rapidement, associée à une dégénérescence des fibres optiques. Dans certains cas moins gravissimes, on peut assister à une transformation de la macula qui devient poivre et sel (ce qui a été noté dans la maladie de Faber).
Une biopsie conjonctivale peut permettre de retrouver la surcharge cellulaire
Traitement
Il n'existe pas de traitement de cette maladie.
Un conseil génétique est possible, grâce au diagnostic prénatal. On peut réaliser un diagnostic pré-implantantoire sur l'embryon pour voir s'il est porteur ou pas de la maladie, après une fécondation in-vitro (FIV). C'est souvent à la suite de cas familiaux qu'on peut entreprendre cette démarche, en respectant les lois de bioéthique.
La recherche est déjà parvenue à traiter le déficit enzymatique de l'équivalent de la maladie de Tay-Sachs, chez la souris "knock-out". Après administration intra-veineuse d'un vecteur viral (adénovirus), les chercheurs ont constaté, dans le sérum, la présence de l'enzyme et l'expression du gène dans les tissus périphériques.
Guidotti JE, Mignon A, Haase G, Caillaud C, McDonell N, Kahn A, Poenaru L. Adenoviral gene therapy of the Tay-Sachs disease in hexosaminidase A-deficient knock-out mice. Hum Mol Genet. 1999 May;8(5):831-8.
Dor Yeshorim
Le Rabbin Josef Eckstein qui avait eu quatre enfants morts de la maladie de Tay-Sachs, et un médecin, ont créé en 1983 à New-York une organisation nommée "Chevra Dor Yeshorim" ("generation of the righteous").
Elle a pour but de détecter les maladies autosomiques récessives mortelles fréquentes dans la communauté des Juifs Ashkénazes : principalement la maladie de Tay Sachs, mais aussi la maladie de Canavan, l'anémie de Fanconi, la dégénérescence fibreuse cystique (cystic fibrosis) et la maladie de Gaucher. Cela se réalise principalement dans les familles ultra-orthodoxes, dont les mariages sont arrangés par les familles, et qui refusent l'avortement quand la vie de la mère n'est pas en danger.
Dor Yeshorim propose donc un tests prénuptial Tay-Sachs au futur couple et a déjà testé plus de 100.000 personnes. Les résultats sont envoyés au rabbin de l'organisation Dor Yeshorim mais pas aux intéressés. Ils ne seront prévenus que s'ils sont tous les deux porteurs du gène défectueux, mais on ne leur dira rien si uniquement l'un des deux est porteur. Les personnes concernées risquent d'avoir à vivre avec ce que le rabbin Shafran appelle «le fardeau affectif d'un savoir, qui ne peut qu'être accablant».
La fréquence de la maladie de Tay-Sachs a nettement diminué chez les Ashkénazes depuis ces tests. Certains n'apprécient pas cette recherche d'anomalies génétiques, comme le rabbin Moshe D. Tendler, qui dit: "You violate my privacy if you know more about me than I know about myself." (Vous violez ma vie privée si vous en savez plus sur moi que moi-même). Ces tests entraînent en effet des problèmes éthiques et des situations familiales difficiles.
Sites web
En français :
En anglais :
Bibliographie
Beutler E. Subunit structure of the hexosaminidase isozymes. Adv Genet. 2001;44:93-100.
Brady RO. Tay-Sachs disease: the search for the enzymatic defect. Adv Genet. 2001;44:51-60.
Burg, J.; Conzelmann, E.; Sandhoff, K.; Solomon, E.; Swallow, D. M. : Mapping of the gene coding for the human GM2 activator protein to chromosome 5. Ann. Hum. Genet. 49: 41-45, 1985.
Chamoles NA, Blanco M, Gaggioli D, Casentini C. Tay-Sachs and Sandhoff diseases: enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnoses in newborn-screening cards. Clin Chim Acta. 2002 Apr;318
Desnick RJ, Kaback MM. Future perspectives for Tay-Sachs disease. Adv Genet. 2001;44:349-56.
Ekstein J, Katzenstein H. The Dor Yeshorim story: community-based carrier screening for Tay-Sachs disease. Adv Genet. 2001;44:297-310.
Guidotti JE, Mignon A, Haase G, Caillaud C, McDonell N, Kahn A, Poenaru L. Adenoviral gene therapy of the Tay-Sachs disease in hexosaminidase A-deficient knock-out mice. Hum Mol Genet. 1999 May;8(5):831-8.
Hechtman, P.; Gordon, B. A.; Ng Ying Kin, N. M. K. : Deficiency of the hexosaminidase A activator protein in a case of GM2 gangliosidosis; variant AB. Pediat. Res. 16: 217-222, 1982.
Hechtman P, Boulay B, De Braekeleer M, Andermann E, Melancon S, Larochelle J, Prevost C, Kaplan F. The intron 7 donor splice site transition: a second Tay-Sachs disease mutation in French Canada. Hum Genet. 1992 Dec;90(4):402-6.
Hill LW, Schorr SJ. Prenatal screening for Tay-Sachs disease by Louisiana obstetricians: a survey study. South Med J. 2001 Sep;94(9):910-2.
McDowell GA, Mules EH, Fabacher P, Shapira E, Blitzer MG. The presence of two different infantile Tay-Sachs disease mutations in a Cajun population. Am J Hum Genet. 1992 Nov;51(5):1071-7.
Martino S, Emiliani C, Tancini B, Severini GM, Chigorno V, Bordignon C, Sonnino S, Orlacchio A. Absence of metabolic cross-correction in TAY-SACHS cells: Implications for gene therapy. J Biol Chem. 2002 Mar 28
Mosenkis A. Genetic screening for breast cancer susceptibility: a Torah perspective. J Halacha Contemporary Society. 1997 Fall;No. 34:5-26.
Okada S, O'Brien JS. Discovery of beta-hexosaminidase A deficiency in Tay-Sachs disease. Adv Genet. 2001;44:61-6.
Palomaki GE, Williams J, Haddow JE, Natowicz MR. Tay-Sachs disease in persons of French-Canadian heritage in northern New England. Am J Med Genet. 1995 May 8;56(4):409-12.
Prence EM, Jerome CA, Triggs-Raine BL, Natowicz MR. Heterozygosity for Tay-Sachs and Sandhoff diseases among Massachusetts residents with French Canadian background. J Med Screen. 1997;4(3):133-6.
Rothenberg KH, Rutkin AB. Toward a framework of mutualism: the Jewish community in genetics research. Community Genet. 1998;1(3):148-53.
Rozenberg R, Pereira Ld. The frequency of Tay-Sachs disease causing mutations in the Brazilian Jewish population justifies a carrier screening program. Sao Paulo Med J. 2001 Jul;119(4):146-9.
Sakuraba, H.; Itoh, K.; Shimmoto, M.; Utsumi, K.; Kase, R.; Hashimoto, Y.; Ozawa, T.; Ohwada, Y.; Imataka, G.; Eguchi, M.; Furukawa, T.; Schepers, U.; Sandhoff, K. : GM2 gangliosidosis AB variant: clinical and biochemical studies of a Japanese patient. Neurology 52: 372-377, 1999.
Sandhoff K. The GM2-gangliosidoses and the elucidation of the beta-hexosaminidase system. Adv Genet. 2001;44:67-91.
Xie, B.; Kennedy, J. L.; McInnes, B.; Auger, D.; Mahuran, D. : Identification of a processed pseudogene related to the functional gene encoding the G-M2 activator protein: localization of the pseudogene to human chromosome 3 and the functional gene to human chromosome 5. Genomics 14: 796-798, 1992.
Zlotogora J, Leventhal A. Screening for genetic disorders among Jews: how should the Tay-Sachs screening program be continued? Isr Med Assoc J. 2000 Sep;2(9):665-7.

![]()
Suivez nous sur...
![]()
SNOF
10 rue Schweighaeuser
CS 40028
67080 STRASBOURG Cedex
Tél. 03 88 35 01 09
Fax. 03 88 25 51 90