Le site des ophtalmologistes de France
Espace Encyclopédie
Encyclopédie de la vue

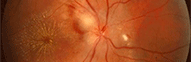




Rétinoschisis juvénile lié à l'X
Rétinoschisis juvénile lié à l'X
X-linked Retinoschisis (XLRS)
Dr Patrice Déglise Toulouse France
- Nous remercions, pour son aide, le Docteur Christian Hamel, Directeur de Recherches et responsable de l'équipe génétique Inserm U. 254 à Montpellier France, ainsi que le Dr Patrice Déglise (Toulouse France).
Définition
Il s'agit d'une affection génétique rare qui atteint, très généralement, les garçons et les jeunes hommes, et qui entraîne une diminution progressive de l'acuité visuelle. La majorité de ces patients est âgée de 10 à 20 ans, mais les nourrissons semblent déjà atteints. Le diagnostic peut ne pas être fait au début car les signes oculaires sont parfois difficiles à percevoir. C'est souvent à l'école qu'on se rend compte des difficultés d'apprentissage.
On voit apparaître des formations kystiques dans la rétine, ce qui va entraîner un clivage rétinien à l'origine des altérations anatomiques (rétinoschisis vient du grec schizein partager, scinder).
La protéine en cause est exprimée dans les photorécepteurs et actuellement on ne comprend pas pourquoi à partir de cette expression dans les photorécepteurs on a un schisis. La participation des cellules de Müller, que certains auteurs ont avancée, reste à l'heure actuelle spéculative.
Historique
Cette maladie a été décrite par Haas en 1898 qui observa au niveau maculaire des kystes en 'rayon de roue' (spoke-wheel pattern), puis par Pagenstecher en 1913 qui montra le caractère familial.
C'est Wilczek qui introduisit le terme de rétinoschisis en 1935.
Ida Mann et MacRae ont décrit en 1938 des "congenital vascular veils in the vitreous".
Génétique
Dans la grande majorité des cas, ce sont les hommes jeunes XmY qui sont atteints car la transmission héréditaire est récessive liée au sexe. La pénétrance est très élevée (95%) et les phénotypes très variés.
Les femmes porteuses du gène malade étant XmX ne sont pas atteintes cliniquement car un de leur X est sain, mais elles transmettent la maladie (elles sont conductrices).
50% de leurs garçons seront atteints (XmY), 50% des garçons seront sains (XY).
50% de leurs filles seront conductrices ( XmX), 50% seront saines (XX).
Il existent de rares femmes homozygotes atteintes (XmXm), lors de mariages consanguins. Elles auront un rétinoschisis juvénile.
Parfois les femmes conductrices XmX ont quelques signes cliniques à cause de la lyonisation. Ce phénomène normal, qui a lieu pendant la phase embryonnaire, correspond à l'inactivation d'un chromosome X, laissant l'autre fonctionnel. Selon la proportion d'Xm non inactivé, on pourra avoir des signes pathologiques plus ou moins marqués chez les femmes.
Ainsi, deux signes sont caractéristiques des femmes conductrices :
- la perte du reflet fovéolaire
- l'irrégularité de la pigmentation maculaire.
Le gène en cause est XLRS1 et se situe sur le chromosome X en position Xp22.2-p22.1. Ce gène a six exons et code une protéine de 224 acides aminés qui contient un domaine discoïdine indispensable pour un développement normal de la rétine. Différentes mutations ont été décrites au niveau de ce gène.
Acuité visuelle
Elle est toujours faible, entre 2 et 4/10ème, souvent meilleure en vision de près (P2 ou 3).
Une hypermétropie fréquente est retrouvée par les auteurs.
Clinique
 |
 |
 |
 |
Clichés Dr Patrice Déglise Toulouse France
Cliquer pour agrandir
Trois types d'altérations forment le tableau clinique:
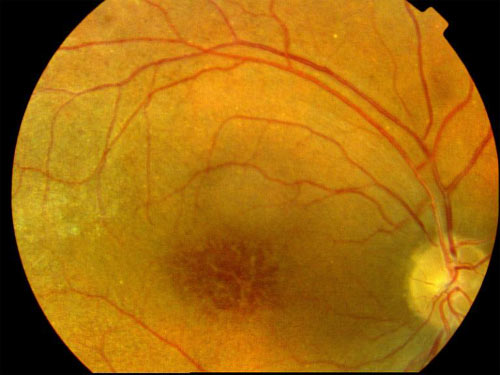
- Une atteinte maculaire bilatérale, encore appelée rétinoschisis central, est responsable de la baisse de vision. Elle se caractérise par la présence de logettes kystoïdes grosses au niveau de la foveola et en étoiles dans la zone périfovéolaire. On note de fines stries périmaculaires. Cela ne s'accompagne pas de diffusion (leakage) ni d'accumulation (staining) lors de l'angiographie, contrairement à l'oedème cystoïde.
La ligne de profil antérieur de la rétine est modifiée et bombée par ses éléments kystiques. Cela correspond à une altération des couches des fibres visuelles ou des cellules de Müller, principales cellules gliales de la rétine. - Des lésions périphériques rétiniennes associent des plages mouchetées, palissadiques ou micacées à un rétinoschisis vrai périphérique. On retrouve celui-ci surtout en temporal inférieur. On peut parfois noter des périvascularites qui vont laisser fuir le colorant à l'angiographie.
Ce soulèvement progresse vers la macula et présente parfois des trous dans son feuillet interne. C'est une des principales causes d'hémorragies du vitré des jeunes hommes. - Des lésions vitréennes à type de voiles bilatéraux, complètent l'ensemble. Cette description correspond à des zones de schisis qui se sont détachées et ont migré dans le vitré.
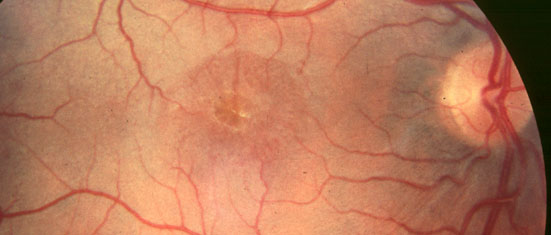
Rétinoschisis juvénile
Cliché Dr Christian Hamel Inserm Montpellier France
Les femmes conductrices de la maladie, hétérozygotes, présentent souvent des altérations de la rétine périphérique qui sont les stigmates de la maladie. On s'attachera à rechercher ces indicateurs génétiques.
Electrophysiologie
Ces examens capitaux sont très parlants.
L'électrorétinogramme (ERG) est toujours perturbé et montre une onde b pathologique peu élevée (couches rétiniennes internes), l'onde a étant normale (photorécepteurs normaux). On parle d'électrorétinogramme électronégatif. Cette onde b plate est sans doute due à un mauvais fonctionnement des cellules de Müller (Mueller cells) qui sont chargées, entre autres fonctions, de gérer l'évacuation du potassium du secteur extra-cellulaire. Dans les formes évoluées tardives, l'onde a devient pathologique.
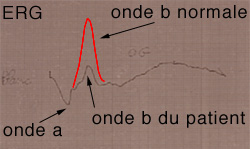
Electrorétinogramme
Onde b réduite
Diagnostic différentiel
- Syndrome de Goldmann-Favre
- Dystrophie vitréo-rétinienne de Wagner
- Rétinoblastome
- Maladie de Stargardt
Evolution
Elle est lente, à moins qu'une complication survienne (rare décollement de rétine par déchirure du feuillet externe, hémorragie par rupture des vaisseaux altérés au niveau des trous du feuillet interne).
On assiste à la transformation de l'aspect maculaire microkystique en aspect macrokystique en 'nid d'abeilles', puis en trou maculaire, puis en atrophie maculaire avec effet fenêtre à l'angiographie. Il est alors souvent difficile de faire le diagnostic devant ce tableau peu spécifique.
On a décrit des néovaisseaux prépapillaires ou prérétiniens, favorisant les hémorragies du vitré.
La vision baisse progressivement à des valeurs de 1/10ème ou 1/20ème vers 50 ou 60 ans.
Les patients ne sont quasiment jamais aveugles et gardent une certaine vision.
Traitement
Il n'existe pas de traitement de cette affection.
On devra tout de même surveiller les patients pour traiter les complications pouvant apparaître (décollement, hémorragie du vitré), ou envisager parfois une photocoagulation laser ou une cryothérapie des lésions périphériques.
Cette chirurgie du décollement de rétine est souvent difficile et peut être aidée par l'utilisation de décaline (perfluorocarbone liquide). L'intervention sur une hémorragie du vitré est souvent discutée, mais peut être envisagée en cas de risque d'amblyopie.
Il faut envisager un examen des membres de la famille pour rechercher des signes pathologiques.
Bibliographie
Arden, G. B.; Gorin, M. B.; Polkinghorne, P. J.; Jay, M.; Bird, A. C. : Detection of the carrier state of X-linked retinoschisis. Am. J. Ophthal. 105: 590-595, 1988.
Barbat V. Particularités de la chirurgie du segment postérieur en cas de rétionoschisis juvénile lié à l'X. Les cahiers d'ophtalmologie.Mars 2002:27-30.
Dahl, N.; Goonewardena, P.; Chotai, J.; Wadelius, C.; Lindsten, J.; Pettersson, U. : DNA linkage analysis of X-chromosome linked retinoschisis. Cytogenet. Cell Genet. 46: 602, 1987.
Gehrig, A.; Weber, B. H. F.; Lorenz, B.; Andrassi, M. : First molecular evidence for a de novo mutation in RS1 (XLRS1) associated with X-linked juvenile retinoschisis. J. Med. Genet. 36: 932-934, 1999.
Haas, J. : Ueber das Zusammenvorkommen von Veranderungen der Retina und Chorioidea. Arch. Augenheilk. 37: 343-348, 1898.
Hamel CP, Griffoin JM, Bazalgette C, Lasquellec L, Duval PA, Bareil C, Beaufrere L, Bonnet S, Eliaou C, Marlhens F, Schmitt-Bernard CF, Tuffery S, Claustres M, Arnaud B. Molecular genetics of pigmentary retinopathies: identification of mutations in CHM, RDS, RHO, RPE65, USH2A and XLRS1 genes J Fr Ophtalmol. 2000 Dec;23(10):985-95.
Kaplan, J.; Pelet, A.; Hentati, H.; Jeanpierre, M.; Briard, M. L.; Journel, H.; Munnich, A.; Dufier, J. L. : Contribution to carrier detection and genetic counselling in X linked retinoschisis. J. Med. Genet. 28: 383-388, 1991.
Khan NW, Jamison JA, Kemp JA, Sieving PA. Analysis of photoreceptor function and inner retinal activity in juvenile X-linked retinoschisis. Vision Res. 2001 Dec;41(28):3931-42.
Koh AH, Hogg CR, Holder GE. The incidence of negative ERG in clinical practice. Doc Ophthalmol. 2001 Jan;102(1):19-30.
Mann I., MacRae A. Congenital vascular veils in the vitreous. Br J Ophthalmol 1938;22:1-10.
Miller MP, Kumar S. Understanding human disease mutations through the use of interspecific genetic variation. Hum Mol Genet. 2001 Oct 1;10(21):2319-28.
Musarella MA. Molecular genetics of macular degeneration. Doc Ophthalmol. 2001 May;102(3):165-77.
Pagenstecher, H. E. : Ueber eine unter dem Bilde der Netzhautablosung verlaufende, erbliche Erkrankung der Retina. Graefes Arch. Ophthal. 86: 457-462, 1913.
Retinoschisis Consortium : Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis (XLRS). Hum. Molec. Genet. 7: 1185-1192, 1998.
Sieving PA, Yashar BM, Ayyagari R. Juvenile retinoschisis: a model for diagnostic testing of x-linked ophthalmic disease. Trans Am ophthalmol soc 1999;97:451-464 Am J Ophthalmol. 2000 Jun;129(6):833.
Springer, W. R.; Cooper, D. N.; Barondes, S. H. : Discoidin I is implicated in cell-substratum attachment and ordered cell migration of Dictyostelium discoideum and resembles fibronectin. Cell 39: 557-564, 1984.
Stanga PE, Chong NH, Reck AC, Hardcastle AJ, Holder GE. Optical coherence tomography and electrophysiology in X-linked juvenile retinoschisis associated with a novel mutation in the XLRS1 gene. Retina. 2001;21(1):78-80.
Van de Vosse, E.; Bergen, A. A. B.; Meershoek, E. J.; Oosterwijk, J. C.; Gregory, S.; Bakker, B.; Weissenbach, J.; Coffey, A. J.; van Ommen, G.-J. B.; Den Dunnen, J. T. : An Xp22.1-p22.2 YAC contig encompassing the disease loci for RS, KFSD, CLS, HYP and RP15: refined localization of RS. Europ. J. Hum. Genet. 4: 101-104, 1996.
Wilczek M Ein der netzhautspaltung (retinoschisis) mit einer offnung. Z Augenheilkd 1935;5:13-17.
Yanoff, M.; Kertesz Rahn, E.; Zimmerman, L. E. : Histopathology of juvenile retinoschisis. Arch. Ophthal. 79: 49-53, 1968.
Sites web
Online Mendelian Inheritance in Man (Site américain de génétique)
Association Nationale de Parents d'Enfants Aveugles
Orphanet Le site des maladies orphelines

![]()
Suivez nous sur...
![]()
SNOF
10 rue Schweighaeuser
CS 40028
67080 STRASBOURG Cedex
Tél. 03 88 35 01 09
Fax. 03 88 25 51 90