Le site des ophtalmologistes de France
Espace Encyclopédie
Encyclopédie de la vue

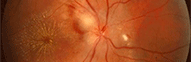




Colobomes
Les colobomes
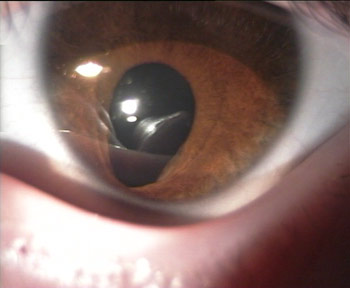
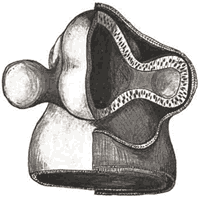
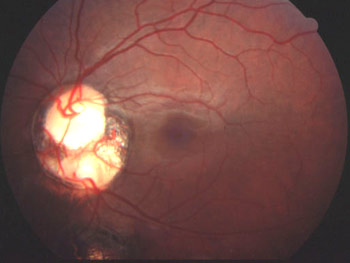
Colobome irien (oeil gauche)
Photo Dr Jean-Michel Muratet
1) Définition
Le terme colobome vient du grec 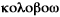 qui veut dire "je mutile".
qui veut dire "je mutile".
La définition de Van Duyse résume cette anomalie oculaire congénitale : "La dénomination de colobome congénital s'applique aux malformations de certaines parties de l'oeil. Leur configuration, leur siège, donnent un aspect caractéristique à ces anomalies. Ce sont des fentes (iris), des lacunes (chorio-rétine), le tissu normal étant absent, aplasique ou remplacé par du tissu connectif; ce sont des modifications de forme (nerf optique) relevant, pour toutes ces anomalies d'un processus pathologique au cours de la vie intra-utérine. Ces malformations siègent d'ordinaire dans le méridien répondant au niveau de la fente foetale, soit directement en bas, ou dans un méridien un peu plus interne (colobomes typiques). Elles peuvent être localisées en d'autres méridiens quelconques (colobomes atypiques)."
Ces anomalies sont dues à un défaut de fermeture de la fissure foetale. Ainsi on peut trouver des fentes qui siègent sur les paupières, la cornée, l'iris, la zonule et le corps ciliaire, la choroïde, la rétine et le nerf optique.
Cette anomalie est souvent associée à une microphtalmie, et peut s'associer à la présence d'un kyste qui va prendre place dans l'orbite. La survenue d'un tel kyste n'est pas bien expliqué, certains auteurs émettant l'hypothèse qu'il est dû à un défaut de sclère.
2) Historique
En 1673 c'est l'anatomiste danois Thomas Bartolin (le jeune) qui décrivit le premier un colobome de l'iris.
En 1821 Von Graefe observe le premier, grâce à l'ophtalmoloscope, un colobome chorio-rétinien typique.
En 1876 Manz émet l'idée que c'est le mésoderme qui joue un rôle important dans la génèse du colobome.
En 1937 Ida Mann étudia de façon précise les anomalies du développement de l'oeil ainsi que son embryologie.
3) Embryologie
Nous ne faisons qu'effleurer le sujet car un chapitre sur l'embryologie de l'oeil est disponible.
Le colobome est dû à un défaut de fermeture de la fente colobomique aux alentours de la 5-7 ème semaine de vie, quand l'embryon a une taille de 7 à 14 mm. C'est la période pendant laquelle on assiste à l'invagination de la vésicule optique et la fermeture de la fente foetale.
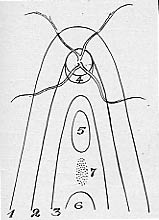
Sur les schémas ci-dessous, on se rend bien compte que l'oeil de l'embryon a la forme d'un bourgeon, puis d'une espèce de cupule qui va se refermer progressivement en bas au niveau de la fente foetale.
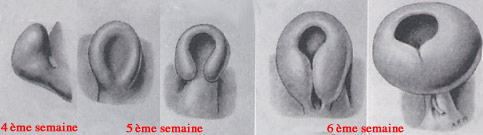
Fermeture de la fente colobomique
Hamilton et al. Human embryology 1972
Rappel
L'oeil se développe à partir de trois couches, le neuro-ectoderme, la crête neurale et l'ectoderme de surface. Le mésoderme ne joue qu'un faible rôle dans la construction de l'oeil car il ne participe qu'à la formation des muscles striés extra-oculaires et l'endothélium vasculaire.
- Le neuro-ectoderme va donner la vésicule optique et la cupule optique. Il va former l'épithélium pigmenté rétinien, les muscles dilatateurs et sphincter de l'iris, l'épithélium iriien, l'épithélium ciliaire, les fibres nerveuses du nerf optique et la glie.
- L'ectoder:me de surface va former le cristallin, l'épithélium cornéen, la glande lacrymale, l'épiderme des paupières, l'épithélium des glandes annexes et de la conjonctive.
- Les crêtes neurales sont à l'origine des kératocytes cornéens, de l'endothélium cornéen et du trabeculum, du stroma irien et chorodien, du muscle ciliaire, des fibroblastes de la sclère, des méninges du nerf optique. Elles donnent aussi le tissu fibroadipeux orbitaire, les nerfs de l'orbite et les cellules de Schwann, le ganglion trigéminé, ainsi que les cartilages et les os orbitaires.
L'ébauche des yeux apparaît vers le 22ème jour, comme 2 sillons de chaque côté de la ligne médiane à l'extrémité encore ouverte des plis neuraux antérieurs. Ces 2 sillons sont situés entre le télencéphale et le diencéphale et sont donc formés par l'invaginatin des plis neuraux. Cela va aboutir à la formation des gouttières optiques qui se transformeront en vésicules optiques. Chaque vésicule est connectée au cerveau par un pédicule qui formera le nerf optique et contient les vaisseaux hyaloidaux.
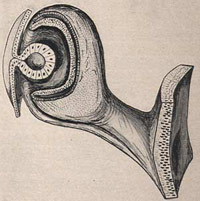
Développement des vésicules optiques
Ida Mann 1928
Au même moment apparaît un épaississement de l'ectoderme de surface qui va former la placode cristallinienne qui va s'invaginer pour former la vésicule cristallinienne (futur cristallin).
Embryologie oculaire
Ida Mann 1928
La vésicule optique va alors aussi s'invaginer donnant une cupule optique à double couche cellulaire. L'invagination simultanée de la vésicule optique et du pédicule optique va aboutir à la création de la fissure foetale, en position inférieure. Les vaisseaux du mésoderme vasculaire poussent alors dans le mésenchyme de la fissure. Ce sont les vaisseaux hyaloidiens qui deviendront plus tard l'artère et la veine centrale de la rétine. Les deux bords de la vésicule optique se joignent petit à petit et vont permettre la fermeture de la fente, progresssivement, pendant la 5ème et 6ème semaine. Tout anomalie de fermeture donnera un colobome.
4) Clinique
Colobome irien
Quand ils sont isolés, ils n'entraînent souvent pas d'anomalie de la vision et n'ont qu'un inconvénient esthétique. C'est l'association avec un colobome chorio-rétinien qui entraîne des perturbations sévères de la vision.
La forme typique la plus classique est le colobome irien inféro-nasal, comme sur la première image de ce chapitre. La gravité vient surtout des anomalies associées, que ce soit un colobome chorio-rétinien et/ou papillaire, ou bien une anomalie oculaire grave et/ou neurologique. En effet, autant un colobome irien peut ne donner aucune conséquence sur la vision, autant un colobome postérieur (chorio-rétinien et/ou papillaire) va souvent s'accompagner d'une faible vision non améliorable.
L'emplacement des colobomes atypiques (en dehors de l'axe inférieur) n'a pas encore été bien expliqué, bien qu'on ait proposé différentes théories.
Un colobome peut être complet et intéresser toute l'épaisseur de l'iris (épithélium et stroma) ou bien être incomplet et ne concerner qu'une partie de l'épaisseur de l'iris.
Un colobome peut être total et entrainer une ouverture jusqu'à la racine de l'iris en "trou de serrure", ou bien partiel et ne donner qu'une déformation de la pupille qui n'est alors plus ronde.
On peut voir persister quelques fibres en travers du colobome, elles correspondent à des fibres mésodermiques pouvant aboutir à la présence de plusieurs (fausses) pupilles, c'est une polycorie.
Colobome cristallinien
Quand l'embryon a une longueur de 26mm le vitré secondaire se modifie et principalement dans sa partie antérieure nommée faisceau isthmique.
Les anatomistes et embryologistes ne sont pas d'accord sur l'origine exacte des fibres zonulaires qui vont suspendre le cristallin. Leur absence en regard d'un segment du cristallin va donner une encoche dans le cristallin. C'est cela qu'on appelle une "colobome" cristallinien bien que ce ne soit pas un vrai colobome car il ne manque pas de substance, il y a juste une rétraction d'un secteur cristallinien. Cette anomalie est souvent isolée et ne s'accompagne pas de perturbations du trabeculum ou de l'angle irido-cornéen, mais parfois d'anomalies du corps ciliaire.

Colobome inféro-nasal du cristallin
Colobome chorio-rétinien
Ce sont là des colobomes graves car responsables d'une mauvaise vision.
Nous avons vu que la fermeture de la fente s'effectuait à la 6ème semaine, date à laquelle les cellules du neuro-épithélium rétinien recouvrent l'intérieur de l'oeil. Cette couche (layer) s'applique sur le feuillet externe rétinien qui constitue l'épithélium pigmenté rétinien. L'absence d'accolement va donner un colobome. La choroide ne pourra pas se différencier normalement si elle ne trouve pas de feuillet pigmenté rétinien. Apparaît alors une zone sans rétine ni choroide fonctionnelle. La sclère blanche sera alors apparente dans cette zone dépouvue de choroide et de rétine. Parfois on peut observer une rétine non différenciée dans cette zone, éventuellement vascularisée. La sclère de cette zone peut être malformée et donner une protrusion postérieure, un staphylome plus ou moins étendu.
L'aspect de ces colobomes chorio-rétiens est protéiforme, car des zones de rétine saine peuvent persister entre les zones d'atrophie chorio-rétinienne. On découvre ce fond d'oeil anormal quand on examine un enfant qui a un colobome irien ou bien si on a été alerté par un reflet blachâtre du fond d'oeil (leucocorie). En principe la localisation est inféro-nasale. Si la lésion est limitée ou bien isolée, il se peut que le diagnostic ne soit fait que très tardivement ou pas du tout.
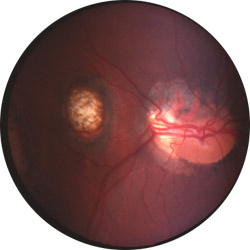 |
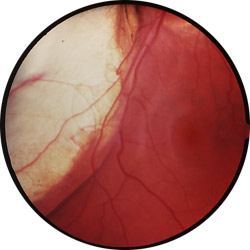 |
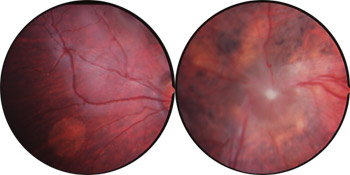
Colobomes chorio-rétiniens
Cliché Pr Mathis CHU Toulouse-Rangueil France
Les colobomes maculaires peuvent être isolés et confondus avec une maladie de Leber, une toxoplasmose ou une toxocarose oculaire, car l'aspect est très proche. Ce sont les examens complémentaires qui pourront faire la différence (électrorétiogramme, sérologies). Ces colobomes sont en principe bilatéraux.
Colobomes du nerf optique
Ces anomalies peuvent être isolées ou associées à des colobomes chorio-rétiniens. En fonction de l'importance du défect tissulaire, on a décrit six types de colobomes papillaires, allant de la petite indentation papillaire à la plus importante anomalie qui en permet pas de reconnaître la papille englobée dans une zone blachâtre de déficit tissulaire majeur. Ceci permet d'essayer de prédire la vision de ces enfants souvent difficiles à examiner et à interroger.
Colobome chorio-rétinien touchant la papille
Parfois peuvent s'associer des encéphalocèles avec hernie de tissu cérébral dans l'ouverture, donnant des exophtalmies ou des tuméfactions palpébrales supérieures. Ils ne faut absolument pas aller biopsier ces tuméfactions de la paupière supérieure car on va au devant de dramatiques problèmes...
Des anomalies neurologiques graves peuvent se retrouver, comme une agénésie du corps calleux, des dysfonctions endocriniennes et des défects de la selle turcique.
Une catégorie spéciale de colobome est le Mornig Glory Syndrome qui est évoqué sur une page particulière.
Ida Mann classait les colobomes en 7 catégories :
|
1 Le colobome englobe la papille et s'étend au dessus d'elle 2 Le colobome englobe la papille mais ne la dépasse pas 3 Ces deux variétés peuvent présenter un pont de tissu normal qui divise le colobome en deux parties, une partie supérieure et une partie inférieure, c'est le colobome à pont 4 Le colobome est localisé au bord inférieur de la papille sous forme d'un croissant 5 Le colobome n'atteint ni la papille ni la périphérie et se présente sous forme d'une tâche blachâtre ovalaire. 6 Le colobome est tout à fait périphérique 7 Le colobome est rudimentaire., il n'existe qu'une trainée pigmentaire située dans la direction de la fente foetale. |
 D'après Ida Mann |
Colobomes associés à une microphtalmie ou un kyste
Une microphtalmie correspond à une oeil plus petit que la normale (à partir de deux déviations standards par rapport à une taille normale). Un globe oculaire a une longueur de 18mm à la naissance. Un anophtalmie correspond à une microphtalmie extrême pour laquelle les structures oculaires ne sont reconnaissables qu'au microscope. Les colobomes chorio-rétiniens peuvent s'accompagner de microphtalmie, le mauvais développement oculaire étant la cause de ces anomalies.
On décrit aussi des kystes oculaires appendus au nerf optique, présentant un mur postérieur scléral et un mur antérieur formé d'une rétine plus ou moins différenciée.
5) Complications
Les colobomes chorio-rétiniens peuvent s'accompagner de décollement de rétine (DR) et/ou de cataracte. Les décollements sont plus souvent acquis que congénitaux. Cela provient souvent des anomalies chorio-rétiniennes mais aussi des anomalies vitréennes car ces yeux sont souvent myopes. Le vitré alors pathologique a tendance à tirer sur la rétine.
On a décrit des décollements associés à des colobomes cristalliniens, mais ce sont surtout les colobomes papillaires qui peuvent donner des DR par communications de l'espace virtuel situé entre les deux feuillets rétiniens et l'espace sous-aragnoidien ou bien l'espace vitréen.
Une association colobome et rétinoblastome doit faire évoquer et rechercher une délétion 13q-.
Des néovascularisations sous-rétiniennes doivent être traitées par laser, en fonction de leur emplacement et de leur importance.
Nous ne ferons que citer les complications cornéeennes (ulcérations) conséquences d'un colobome palpébral supérieur.
6) Etiologies
a) Sans anomalie systémique associée
Transmission autosomique dominante
Des colobomes sans associations systémiques sont souvent décrits. Les parents doivent être examinés très méticuleusement à la recherche de minimes anomalies. Dans une famille présentant une telle perturbation génétique, on a calculé qu'un individu sans anomalie a 8.6% de chance d'avoir un enfant avec colobome.
Les colobomes maculaires rentrent aussi dans ce groupe.
Transmission autosomique récessive
Ce groupe est semble-t-il plus rare mais il y a peu d'études.
b) Avec anomalies systémiques associées
Transmission autosomique dominante
Un tel syndrome a été décrit, associant une microphtalmie avec colobome à des carcinomes, des kystes de la mâchoire, des anomalies des côtes et de la colonne vertébrale et de la hanche, et parfois un retard mental.
Un autre syndrome associe une arachnodactylie, un aspect marfanoïde et un colobome uvéal. Le gène déficient a été repéré sur le chromosome 5 et entraîne un tissu connectif de mauvaise qualité.
Transmission autosomique récessive
Nous citerons simplement:
Le syndrome de Meckel-Gruber qui ajoute une microcéphalie, un encéphalocèle occipital, des anomalies cardiaques, une polydactylie, des fissures faciales, et des anomalies du foie et du pancréas à une microphtalmie avec colobome. Le phénotype est semblable à celui de la trisomie 13, habituellement mortel en quelques semaines.
Le syndrome de Sjogren-Larson qui associe un retard mental à la microphtalmie et aux colobomes.
Transmission liée au chromosome X
Le syndrome de Lenz est décrit comme l'association d'une microphtalmie avec ou sans colobome. Il est transmis par les femmes qui n'ont aucun phénotype particulier en dehors parfois d'une cataracte précoce. Les hommes sont des patients petits, avec retards mental, microcéphalie, anomalies des oreilles et des dents.
Le syndrome d'Aicardi ajoute un colobome irien et/ou papillaire, à des anomalies vertébrales, une microcéphalie avec agénésie du corps calleux et un retard mental. La papille est souvent hypoplasique, colobomateuse et entourée de lacunes chorio-rétiniennes. Le syndrome est letal pour les garçons.
Syndrome d'Aicardi
Clichés Pr André Mathis CHU Rangueil-Toulouse FranceLe syndrome Midas (Microphthalmia, derma aplasia, sclerocornea) met en évidence une microphatmie bilatérale avec blépharophimosis et microcéphalie. Le gène en cause est Xp22.3
Le syndrome Catel-Manzke est caractérisé par des anomalies des doigts.
Transmission inconnue
On place ici le syndrome CHARGE déjà évoqué dans le Morning Glory Syndrome. Cet acronyme correspond à
- Colobomatous microphthalmia,
- Heart defects,
- Atresia of the choanae,
- Retarded growth and development,
- Genital anomalies and hypoplasia,
- Ear anomalies.
Aberrations chromosomiques
On décrit ici de nombreuses anomalies: la triploidie, les trisomies 13, 18 et 22, les duplications 4q+, 7q+, 9p+, 9p+q+, 13q+, 14q+, 22q+, les délétions 2p22, 4p-, 4del(q12q12.1), 4r, 5(q15q22), Dq-, Dr, 11q-, 13q-, 13r-, 18q-, 18r, des inversions péricentriques Inv(6)(p23q23.1) et une anomalie XY, XYY.
7) Traitements
On traite chirurgicalement les colobomes palpébraux qui risquent de donner des ulcères cornéens.
Comme les colobomes iriens ne donnent pas de trouble visuel, on n'y touchera pas, à moins qur'il faille opérer l'oeil pour une cataracte ou une greffe de cornée. Le port de lentilles cornéennes cosmétiques ou correctrices peut être envisagé.
Les colobomes chorio-rétiniens ne peuvent pas être améliorés, on peut seulement opérer les décollements de rétine associés. Ce sont souvent des yeux fragiles qui rendent toute chirurgie délicate.
Un bilan global sera réalisé pour rechercher des anomalies associées, que ce soit des troubles neurologiques, des malformations à distance ou des dysfonctionnements.
8) Conclusion
Les colobomes sont des anomalies qui peuvent être anodines ou bien être associées à des anomalies organiques sévères. Ces enfants seront examinés conjointement par les pédiatres et les ophtalmos pour déceler tout syndrome qui nécessiterait une prise en charge complexe.
9) Bibliographie
Angra SK, Gupta S, Dada VK, Gupta AK. Coloboma of lens. Indian J Ophthalmol. 1984 Jan-Feb;32(1):21-2.
Ascaso FJ, Del Buey MA, Huerva V, Latre B, Palomar A. Noonan's syndrome with keratoconus and optic disc coloboma. Eur J Ophthalmol. 1993 Apr-Jun;3(2):101-3.
Bavbek T, Ogut MS, Kazokoglu H. Congenital lens coloboma and associated pathologies. Doc Ophthalmol. 1993;83(4):313-22.
Connolly WE, Polomeno RC. Optic disc colobomas. Can J Ophthalmol. 1983 Oct;18(6):299-301.
De Souza CF, Berbigier GA, Costa F, Ruschel SP, Silva T, Schuler L. Optic nerve coloboma in Down syndrome. Clin Dysmorphol. 1995 Apr;4(2):176-7.
Ge N, Crandall BF, Shuler JD, Bateman JB. Coloboma associated with Rubinstein-Taybi syndrome. J Pediatr Ophthalmol Strabismus. 1995 Jul-Aug;32(4):266-8.
Giuffre G. Colobomas of the optic area. MPS. 1989;12(4):100-2.
Gopal L, Badrinath SS, Kumar KS, Doshi G, Biswas N. Optic disc in fundus coloboma. Ophthalmology. 1996 Dec;103(12):2120-6; discussion 2126-7.
Jacobs M, Taylor D. The systemic and genetic significance of congenital optic disc anomalies. Eye. 1991;5 ( Pt 4):470-5.
Magli A, Greco A, Alfieri MC, Pignalosa B. Hereditary colobomatous anomalies of the optic nerve head. Ophthalmic Paediatr Genet. 1986 Aug;7(2):127-30.
Merlob P, Horev G, Kremer I, Nissenkorn I. Morning Glory fundus anomaly, coloboma of the optic nerve, porencephaly and hydronephrosis in a newborn infant: MCPH entity.
Murphy BL, Griffin JF. Optic nerve coloboma (morning glory syndrome): CT findings.
Muratet J. Les colobomes chorio-rétiniens. Thèse de Médecine Toulouse 1957
Olsen TW, Summers CG, Knobloch WH. Predicting visual acuity in children with colobomas involving the optic nerve. J Pediatr Ophthalmol Strabismus. 1996 Jan-Feb;33(1):47-51.
Onwochei B.C., Simon J.W, Bateman J.B., Couture K.C., BS, Mir E. Ocular colobamata Survey of ophthalmology Vol 45, num 3, nov-dec 2000.
Pyhtinen J, Lindholm EL. Imaging in optic nerve coloboma. Neuroradiology. 1996 Feb;38(2):171-4.
Recupero SM, Lepore GF, Plateroti R, Abdolrahimzadeh S. Optic nerve aplasia associated with macular 'atypical coloboma'. Acta Ophthalmol (Copenh). 1994 Dec;72(6):768-70.
Sanyanusin P, McNoe LA, Sullivan MJ, Weaver RG, Eccles MR. OMIM Mutation of PAX2 in two siblings with renal-coloboma syndrome. Hum Mol Genet. 1995 Nov;4(11):2183-4. No abstract available.
Shin DH. Repair of iris coloboma by a closed chamber technique. Ophthalmic Surg. 1990 Dec;21(12):867.
Steahly LP. Retinochoroidal coloboma: varieties of clinical presentations. Ann Ophthalmol. 1990 Jan;22(1):9-14.
Theodossiadis GP, Damanakis AG, Theodossiadis PG. Coloboma of the optic disk associated with retinal vascular abnormalities. Am J Ophthalmol. 1995 Dec;120(6):798-800.

![]()
Suivez nous sur...
![]()
SNOF
10 rue Schweighaeuser
CS 40028
67080 STRASBOURG Cedex
Tél. 03 88 35 01 09
Fax. 03 88 25 51 90