Le site des ophtalmologistes de France
Espace Encyclopédie
Encyclopédie de la vue





Maladie de Recklinghausen
Les phacomatoses
Introduction
Les phacomatoses regroupent différentes maladies :
- la neurofibromatose NF1 de Von Recklinghausen (ci-dessous)
- la neurofibromatose NF2
- la sclérose tubéreuse de Bourneville
- le syndrome de Sturge-Weber-Krabbe
- la maladie de Von Hippel-Lindau
- et diverses dysembryoplasies neuro-ectodermiques.
Leur origine est en effet un dysfonctionnement du tissu ectodermique embryonnaire qui formera la peau, le système nerveux et l'oeil. Ces trois éléments seront donc atteints à des degrés divers dans toutes ces maladies.
La maladie de Recklinghausen
ou neurofibromatose de type 1 (NF1)
 |
 |
 |
Avecl'aimable autorisation du Professeur Bonafé
Service de Dermatologie CHU Toulouse-Rangueil France
1) Généralités
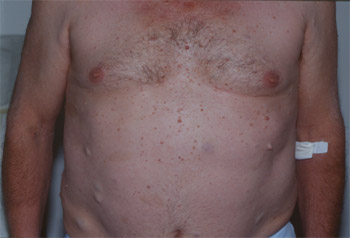
La maladie de Recklinghausen est la plus fréquente des phacomatoses avec un cas sur 3000 naissances et elle se révèle souvent à l'adolescence. Elle correspond à la neurofibromatose de type I (NF1), soit 90% des cas, et elle est due à une anomalie du chromosome 17. On décrit également une forme de type II (NF2) beaucoup plus rare, qui est due à une anomalie du chromosome 22.
La transmission est de type autosomique dominant, caractérisée par le développement de nombreuses tumeurs réparties sur tout le corps. Il s'agit principalement de taches café-au-lait (TCL), associées à des neurofibromes cutanés et sous-cutanés et à des hamartomes iriens.
La pénétrance est très forte, soit près de 100% et la variabilité phénotypique est importante. On note donc de nombreuses formes cliniques, des plus bénignes aux plus graves.
On a évoqué cette maladie pour expliquer la pathologie du patient nommé "Elephant man", Joseph Merrick, mais certains auteurs pensent qu'il s'agissait plutôt d'un syndrome Proteus.
2) Historique
Akenside semble être le premier à avoir décrit un malade porteur de nombreuses verrues qui parsemaient son corps. Cette anomalie avait été héritée de son père.
Tilesius von Tilenau évoque un malade du Professeur Ludwig qui avait écrit à son sujet un texte "Case History of Extraordinarily Unsightly Skin". Ce patient portait également de nombreuses formations cutanées.
Von Recklinghausen, en 1882, décrivit cette maladie qui garda son nom.
3) Atteintes non oculaires
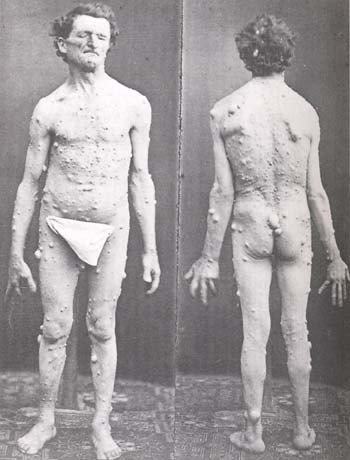
Illustration originale de Von Recklinghausen en 1882
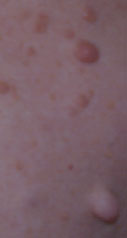
a) Les taches café-au-lait (TCL) sont réparties sur le corps et orientent le diagnostic très jeune. Pour être diagnostiqué Recklinghausen, il faut qu'il y ait un minimum de six taches mesurant au moins 1,5 cm chez l'adulte et 0,5 cm chez l'enfant. Elles sont présentes chez 99% des malades présentant une NF1,et peuvent avoir une dimension variable, de quelques millimètres à plus de 50 cm de diamètre. La biopsie de ces taches met en évidence une grande densité de cellules géantes contenant du pigment (mélanine). Le nombre de mélanocytes est normal. On ne connaît pas l'étiologie de ces TCL et on ne sait pas les enlever.
La présence de TCL sans dysmorphie faciale peut correspondre à une NF1, alors que la présence d'une dysmorphie faciale oriente plutôt vers des syndromes, comme le syndrome de Noonan, le syndrome de Watson ou le syndrome léopard.
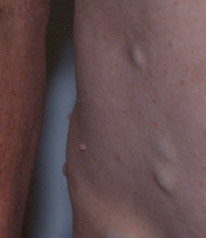
b) Les neurofibromes sont de quatre types :
- Les neurofibromes cutanés peuvent être plus ou moins nombreux. Ils se développent souvent en début de puberté, sont souvent sessiles au début mais deviennent habituellement pédiculés.
- les neurofibromes sous-cutanés sont présents sur les trajets des nerfs
- les névromes plexiformes correspondent à des tumeurs flasques semblant contenir, à la palpation, des "paquets de ficelle".
- les neurofibromes plexiformes diffus
On peut parfois assister à une malignisation des neurofibromes profonds. Le risque de neurofibrosarcome est de 5% environ. Les signes de cancérisation sont l'apparition locale d'une douleur, une augmentation de taille et une insensibilité locale.
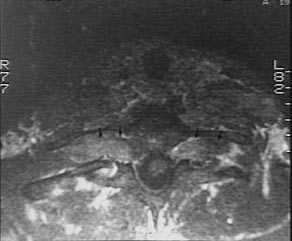
IRM de la moelle cervicale
Neurofibromes dans les trous de conjugaison des vertèbres
Avec l'aimable autorisation du
Collège des enseignants de radiologie de France
c) Les lentigines
Ce sont de petits tâches brunâtres, situés sous les aisselles, qui ressemblent à des tâches café au lait en réduction, mesurant moins de 3 mm de diamètre.
d) Les complications neurologiques
Un retard mental plus ou moins important peut parfois être constaté.
Des maux de tête, un prurit et des douleurs abdominales dues aux neurofibromes intestinaux sont décrits. L'IRM met parfois en évidence des spots brillants correspondant peut-être à une dysplasie gliale et une myélinisation aberrante.
4) Atteintes oculaires
a) Uvée
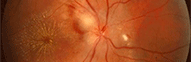
- Les nodules de Sakurai-Lisch
Ils sont pathognomonique de l'affection et correspondent à des mélanocytes groupés avec des cellules gliales. On les voit dans 95% des cas.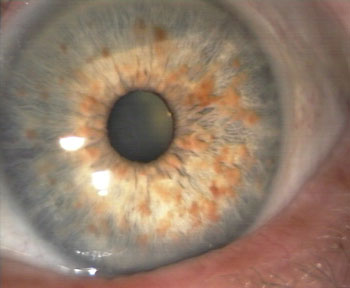
Nodules de Lisch sur l'iris
Cliché Dr Jean-Michel Muratet - Des hamartomes peuvent être vus, issus de la choroide. Au niveau rétinien on n'en voit que rarement, contrairement à la sclérose tubéreuse de Bourneville.
b) Le névrome plexiforme de la paupière supérieure
Cette tumeur unilatérale du bord palpébral provoque un ptosis et présente les mêmes caractères de 'pelotte de ficelle' que les autres névromes. Dans la moitié des cas on constate alors un glaucome du même côté, sans qu'on ne sache pas trop quel mécanisme intervient.
c) Le gliome du nerf optique
Cette tumeur présente sur le nerf optique va entrainer souvent la perte de la fonction visuelle de l'oeil atteint.
Avec l'aimable autorisation du collège des enseignants de radiologie de France
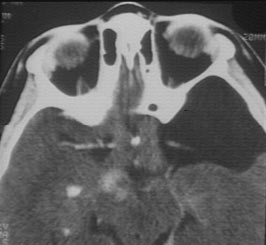 Scanner céphalique Augmentation de taille du chiasma Calcification et prise de contraste anormale de la voie optique droite Kyste arachnoidien |
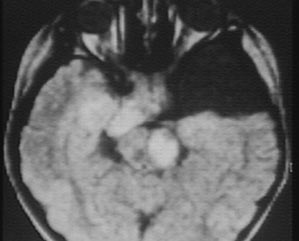 IRM céphalique Même lésions visibles Plus un hypersignal à la partie gauche du pédoncule cérébral |
d) Autres pathologies
On a décrit un épaississement des nerfs intracornéens, des anomalies de l'angle irido-cornéen, un hémangiome rétinien.
Une scoliose importante peut s'ajouter et gêner l'enfant.
5) Diagnostic
Les patients peuvent être considérés comme porteur d'un Recklinghausen quand ils ont au moins deux signes parmi ceux-ci :
- au moins six taches (>1,5 cm après la puberté et 0,5 cm avant la puberté)
- au moins deux neurofibromes,
- un névrome plexiforme de la paupière,
- au moins deux nodules iriens de Lisch,
- un gliome optique
- des lésions osseuses de type dysplasie
- des taches lentigineuses de la région inguinale ou axillaire
- des antécédents directs de neurofibromatose de type I
Le bilan sera surtout neurologique avec un examen clinique complet, des radiographies, scanner et irm céphaliques. On s'attachera à rechercher une hypertension artérielle car les patients sont prédisposés au phéochromocytome.
La réalisation d'une IRM chez l'enfant à la recherche d'un gliome est à discuter avec la famille. Cet examen doit se faire avec prémédication pour les petits enfants qui ne doivent pas bouger du tout. Si on trouve un gliome, la plupart des auteurs préfèrent ne pas y toucher s'il n'entraine aucun symptome.
Un élément important est la réalisation d'un champ visuel, dès que l'enfant peut comprendre le principe de l'examen. Cette surveillance doit se faire régulièrement, au moins chaque année.
7) Génétique
Dès 1987 le gène concerné dans la NFI fut cartographié en 17q11.2 par analyse chromosomique de patients porteurs de la maladie. En 1991 on publia la séquence complète du gène malade. La protéine issue de ce gène fut nommée 'neurofibromine'.
Le gène NF1 est très grand, 300 kb, ce qui favorise les mutations. Il contient au moins 56 exons qui vont donner, après transcription, un ARN messager de 13kb, avec un région codante de 9kb. Les 8454 nucléotides sont à l'origine d'une protéine de 2818 acides aminés. Cette neurofibromine est présente dans de nombreux endroits de l'organisme, mais principalement dans les neurones, les cellules de Schwann et les oligodendrocytes.
Cette protéine semble servir principalement à la croissance et à la régulation du cycle cellulaire et peut-être joue-t-elle un rôle dans la transmission du signal. Elle agit ainsi sur la différenciation cellulaire et la régulation des oncogènes cellulaires.
Cette neurofibromine est associée aux microtubules, ce qui semble être un élément important mais mal élucidé.
On a décrit trois isoformes de la protéine.
8) Le traitement
Les neurofibromes pourront être enlevés s'ils sont trop inesthétiques ou gênants. Ce sont des tumeurs qui saignent facilement et qui sont parfois difficiles à enlever. Leur exérèse sera fonction de leur taille.
Le glaucome dépisté sera souvent opéré.
Un traitement anti-comitial sera éventuellement donné en cas d'épilepsie.
Il n'y a pas de dépistage prénatal (DPN).
9) Les associations
En français :
Association Neurofibromatoses et Recklinghausen
En anglais :
The National Neurofibromatosis foundation
10) Brève bibliographie
Von Recklinghausen, F. : Ueber die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen. Berlin: August Hirschwald (pub.) 1882.
Abeliovich, D.; Gelman-Kohan, Z.; Silverstein, S.; Lerer, I.; Chemke, J.; Merlin, S.; Zlotogora, J. : Familial cafe au lait spots: a variant of neurofibromatosis type 1. J. Med. Genet. 32: 985-986, 1995.
Ars, E.; Serra, E.; Garcia, J.; Kruyer, H.; Gaona, A.; Lazaro, C.; Estivill, X. : Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum. Molec. Genet. 9: 237-247, 2000.
Barker, D.; Wright, E.; Nguyen, K.; Cannon, L.; Fain, P.; Goldgar, D.; Bishop, D. T.; Carey, J.; Baty, B.; Kivlin, J.; Willard, H.; Waye, J. S.; Greig, G.; Leinwand, L.; Nakamura, Y.; O'Connell, P.; Leppert, M.; Lalouel, J.-M.; White, R.; Skolnick, M. : Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science 236: 1100-1102, 1987.
Boudin, G.; Pepin, B.; Vernant, C. : Les tumeurs multiples du systeme nerveux au cours de la maladie de Recklinghausen: à propos d'une observation anatomo-clinique avec adénome chromophobe de l'hypophyse. Presse Med. 78: 1427-1430, 1970.
Cohen, M. M., Jr. : The Elephant Man did not have neurofibromatosis. Proc. Greenwood Genet. Center 6: 187-192, 1987.
Cohen, M. M., Jr. : Understanding Proteus syndrome, unmasking the Elephant Man, and stemming elephant fever. Neurofibromatosis 1: 260-280, 1988.
Cohen, M. M., Jr. : Further diagnostic thoughts about the Elephant Man. Am. J. Med. Genet. 29: 777-782, 1988.
DeClue, J. E.; Cohen, B. D.; Lowy, D. R. : Identification and characterization of the neurofibromatosis type 1 protein product. Proc. Nat. Acad. Sci. 88: 9914-9918, 1991.
Fountain, J. W.; Wallace, M. R.; Brereton, A. M.; O'Connell, P.; White, R. L.; Rich, D. C.; Ledbetter, D. H.; Leach, R. J.; Fournier, R. E. K.; Menon, A. G.; Gusella, J. F.; Barker, D.; Stephens, K.; Collins, F. S. : Physical mapping of the von Recklinghausen neurofibromatosis region on chromosome 17. Am. J. Hum. Genet. 44: 58-67, 1989.
Goldgar, D. E.; Green, P.; Parry, D. M.; Mulvihill, J. J. : Multipoint linkage analysis in neurofibromatosis type 1: an international collaboration. Am. J. Hum. Genet. 44: 6-12, 1989.
Heim, R. A.; Silverman, L. M.; Farber, R. A.; Kam-Morgan, L. N. W.; Luce, M. C. : Screening for truncated NF1 proteins. (Letter) Nature Genet. 8: 218-219, 1994.
Howell, M.; Ford, P. : The True History of the Elephant Man. New York: Penguin (pub.) 1980.
Krone, W.; Hogemann, I. : Cell culture studies on neurofibromatosis (von Recklinghausen): V. Monosomy 22 and other chromosomal anomalies in cultures from peripheral neurofibromas. Hum. Genet. 74: 453-455, 1986.
Riccardi, V. M. : Von Recklinghausen neurofibromatosis. New Eng. J. Med. 305: 1617-1626, 1981.
Upadhyaya, M.; Cooper, D. N. (eds.) : Neurofibromatosis Type 1 from Genotype to Phenotype. Oxford: BIOS Scientific Publ. , 1998.
Zehavi, C.; Romano, A.; Goodman, R. M. : Iris (Lisch) nodules in neurofibromatosis. Clin. Genet. 29: 51-55, 1986.

![]()
Suivez nous sur...
![]()
SNOF
10 rue Schweighaeuser
CS 40028
67080 STRASBOURG Cedex
Tél. 03 88 35 01 09
Fax. 03 88 25 51 90