Le site des ophtalmologistes de France
Espace Encyclopédie
Encyclopédie de la vue





Homocystinurie
L'homocystinurie
Définition
L'homocystinurie a été décrite en 1962 par Carson et Neill. Il s'agit d'une maladie génétique rare (moins d'une personne sur 2000); qui peut conduire à une altération grave de l'état clinique du patient si un traitement (pyridoxine ou vitamine B6) et/ou un régime alimentaire ne sont pas rapidement mis en oeuvre.
Cette pathologie associe une ectopie cristallinienne (un déplacement spontané des cristallins), des occlusions vasculaires, une ostéoporose et parfois des altérations neurologiques.
Elle est due à un déficit d'une enzyme, la cystathionine bêta-synthase (CBS), qui va provoquer l'accumulation (thésaurismose) dans l'organisme de produits toxiques que sont l'homocystéine et la méthionine.
Génétique
Le gène qui est responsable du déficit en CBS, se trouve sur le chromosome 21, en position sous-télomérique 21q22.3.
Il existe une autre forme d'homocystinurie, plus rare encore, qui est due à un déficit en méthyl-tétra-hydrofolate réductase (MTHFR).
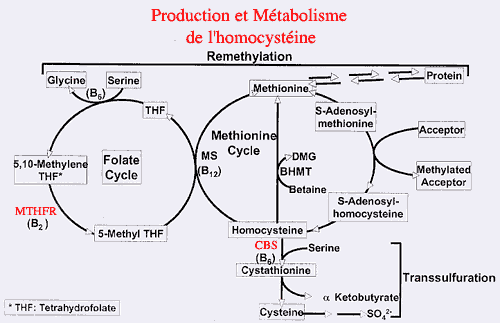
Métabolisme de l'homocystéine
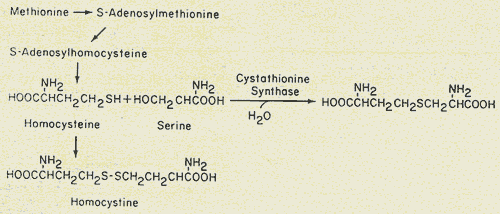
Action de la CBS
La transmission de l'homocystinurie est autosomique récessive.
Sa fréquence dans la population se situe entre 1/60.000 et 1/300.000 personnes, avec des variations entre les pays (1/50.000 en Irlande et 1/1.000.000 au Japon). On note que les patients ont une activité enzymatique entre 0 et 10%, ceux qui sont améliorés par la pyridoxine gardant une petite activité enzymatique qui augmente avec le traitement.
C'est la deuxième encéphalopathie métabolique par ordre de fréquence, après la phénylcétonurie.
L'enzyme CBS est un tétramère de 63kD qui possède des sites d'attache pour le pyridoxal phosphate. L'ADNc pour cet enzyme a été cloné, et le segment concerné comporte 17 exons de 20 à 25 kD. On a découvert plus de 18 mutations. Il n'y a pas de rapport direct entre l'importance des mutations et l'aspect phénotypique.
La déficience de l'enzyme entraîne une forte augmentation de l'homocystinémie et donc une importante élimination urinaire, une forte homocystinurie. Le diagnostic sera fait par l'électrophorès des acides aminés des urines qui retrouvera le pic d'homocystéine, et le dosage de l'homocystine totale.
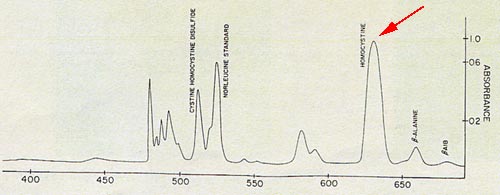
Electrophorèse : Pic d'homocystine
Clinique
1) Lésions oculaires
L'ectopie cristallinienne est un signe important de la maladie et extrêmement fréquent (90% des cas). L'examen ophtalmologique met en évidence deux cristallins déplacés vers le bas. En effet le cristallin est normalement tenu par des fibres, nommées zonule de Zinn, qui l'amarrent au corps ciliaire. Dans la maladie, des fibres se rompent et le cristallin va donc descendre sous l'effet de la pesanteur, dans le plan frontal. La réfraction se modifie alors régulièrement et l'enfant devient de plus en plus myope car la rupture des fibres zonulaires va entraîner un changement de forme du cristallin, qui va devenir plus globuleux et donc plus convergent. L'image se formera plus en avant de la rétine, expliquant la myopie progressive. Malgré une correction optique, le mauvais emplacement des cristallins va aboutir à une vision faible, autour de 1/10ème.
Une ectopie et une myopie doivent faire recherche la maladie.
Cette ectopie peut entraîner un iridodonésis (l'iris bouge avec les mouvements de l'oeil, comme s'il flottait).
Streeten (1982) a remarqué que les fibres zonulaires étaient composées de glycoprotéines qui contiennent une forte concentration de cystéine, ce qui explique très certainement leurs anomalies dans le cadre de cette maladie et la conséquence, l'ectopie cristallinienne.
Les deux complications possibles sont la luxation du cristallin dans le vitré avec les risques de glaucome et décollement de rétine, et la luxation antérieure, dans la chambre antérieure, qui va donner une hypertonie aiguë avec nécessité de chirurgie rapide. Cette intervention chirurgicale va être difficile et présenter des risques majeurs, comme nous le verrons.
Préventivement, il faudra opérer ces enfants quand l'équateur du cristallin sera au niveau de l'axe visuel.
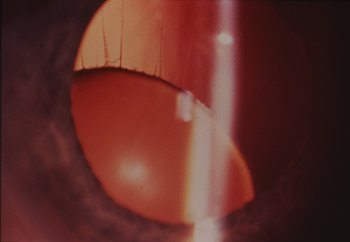
Luxation du cristallin
La rétine présente parfois des altérations de la périphérie (trous ou déchirures) pouvant être responsables de décollement de rétine. Un laser préventif est très difficile à réaliser chez ces enfants présentant souvent un retard mental. Une intervention chirurgicale pour décollement de rétine sera souvent nécessaire, en ayant toujours soin de prendre toutes les précautions pré-opératoires.
Un glaucome peut parfois être observé; la pression intra-oculaire devra donc être évaluée régulièrement.
Le Pr Duffier conclut: "Une surveillance régulière pour ne pas se trouver pris en urgence, une préparation métabolique soignée et une chirurgie quand c'est nécessaire par des spécialistes qui en ont l'habitude sont les trois conditions de succès, en tout cas de prudence vis à vis du traitement opthalmologique des manifestations oculaires de l'homocystinurie."
2) Lésions vasculaires
Ce sont elles qui font la gravité de la maladie. Les vaisseaux artériels et veineux ont en effet tendance à se boucher, à se thromboser, ce qui peut être fatal pour le patient. On décrit des thromboses cérébrales, cardiaques (infarctus) et périphériques (membres) et des embolies pulmonaires.
Le problème peut être dramatique lors d'une intervention chirurgicale car on peut assister à une occlusion très rapide d'une veine ou d'une artère. En effet, la dégénérescence des vaisseaux et la perturbation de leur intima va aboutir à un caillot et une ischémie immédiate. Il faut donc connaître l'homocystinurie avant d'opérer ces patients pour les préparer, car une méconnaissance de la maladie risque d'aboutir à la perte d'une jambe par exemple, quand ce n'est pas au décès du patient. De telles complications sont déjà survenues. L'anesthésiste-réanimateur doit donc bien s'entourer de protocoles anesthésiques particuliers et d'une préparation métabolique.
3) Lésions squelettiques
On peut noter un genu valgum, un thorax en carène ou en creux, et éventuellement une grande taille, un aspect marfanoïde (d'où le risque de se tromper) et une arachnodactylie.
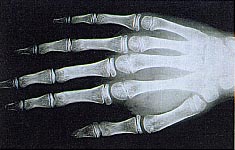
Arachnodactylie
L'ostéoporose est souvent présente et 50% des patients en sont atteints à 30 ans. Des perturbations de la colonne vertébrales sont aussi fréquemment retrouvées.
4) Troubles neurologiques
Ils sont sans doute dus à d'éventuelles perturbations vasculaires intracérébrales. On peut voir un retard mental, des convulsions, des hémiplégies transitoires ou non, éventuellement une dystonie avec torticolis spasmodique.
Des troubles psychiatriques sont parfois décrits (schizophrénie, dépression), et il est donc là important de faire le diagnostic pour soigner les patients malades.
5) Autres lésions
On peut voir une dépigmentation de la peau ou des cheveux. Les extrémités sont souvent froides à cause des troubles vasculaires.
Diagnostic différentiel
La maladie de Marfan est LE diagnostic différentiel qui ne doit pas égarer le diagnostic. Les conséquences peuvent être dramatiques. Dans le Marfan l'ectopie cristallinienne est supérieure ou externe, alors quelle est inférieure dans l'homocystinurie.
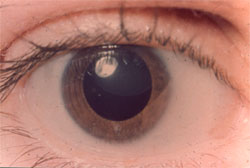
Maladie de Marfan
Ectopie cristallinienne supérieure
Traitement
Un diagnostic prénatal a pu être réalisé par culture d'amniocytes et de villosités choriales, et mise en évidence du déficit en CBS.
Le traitement est la pyridoxine (la vitamine B6) qui est le cofacteur de l'homocystéine et qui améliore nettement un tiers des patients. Tous ne sont pas répondeurs. Un traitement dès le plus jeune âge améliorera bien l'état clinique, d'où l'importance d'un diagnostic précoce.
La posologie est fonction des personnes et correspond à plusieurs centaines de milligrammes par jour.
Des règles diététiques peuvent aussi améliorer ces enfants qui ont des formes résistantes à la pyridoxine. Ce sont des régimes hypoprotidiques plus ou moins strict supplémentés avec un mélange d'acides aminés dépourvu de méthionine et un traitement par la bétaïne.
Une forme particulière
Il existe un type d'homocystinurie par trouble de la reméthylation, à cause d'un déficit en méthyl-tétra-hydrofolate réductase (MTHFR). Ce déficit autosomique récessif empêche la réduction du 5-10 méthylène tétra hydrofolate en 5 méthyl-tétra-hydrofolate, donneur de méthyl permettant la reméthylation de l'homocystéine en méthionine (voir le schéma).
Il y a donc carence en méthyl-tétra-hydrofolate et homocystinurie avec hypométhioninémie.
L'enfant dès sa première années présente des convulsions, une microcéphalie et des apnées récurrentes.
L'affection peut commencer à l'âge adulte et s'accompagner de troubles psychiatriques (schizophrénie, AVC...). Le diagnostic sera fait comme précedemment par chromatographie et dosage de l'homocystine plasmatique. On notera en outre un taux bas de méthyl-folate plasmatique.
Le traitement sera à base de fortes doses de bétaïne associées à un complément en méthionine, en pyridoxine, vitamine B12, acide folique et carnitine.
Adresses internet
Les Enfants du Jardin regroupe les familles d'enfants présentant ces pathologies et soumis à des régimes pauvres en protides.

OMIM (Online Mendelian Inheritance in Man) Le site de référence sur toutes les maladies génétiques HOMOCYSTINURIA (en anglais)
Bibliographie succinte
Blika S, Saunte E, Lunde H, Gjessing LR, Ringvold A. Homocystinuria treated with pyridoxine. Acta Ophthalmol (Copenh). 1982 Dec;60(6):894-906.
Boers, G. H. J.; Smals, A. G. H.; Trijbels, F. J. M.; Fowler, B.; Bakkeren, J. A. J. M.; Schoonderwaldt, H. C.; Kleijer, W. J.; Kloppenborg, P. W. C. : Heterozygosity for homocystinuria in premature peripheral and cerebral occlusive arterial disease. New Eng. J. Med. 313: 709-715, 1985.
Brunova B. [Eye manifestations in homocystinuria]. Cesk Oftalmol. 1987 Jul;43(4):267-70.
Burke JP, O'Keefe M, Bowell R, Naughten ER. Ocular complications in homocystinuria--early and late treated. Br J Ophthalmol. 1989 Jun;73(6):427-31.
Carson, N. A. J.; Neill, D. W. : Metabolic abnormalities detected in a survey of mentally backward individuals in Northern Ireland. Arch. Dis. Child. 37: 505-513, 1962.
Carson NA. Homocystinuria. Proc R Soc Med. 1970 Jan;63(1):41-3.
Carson NA. Homocystinuria. Trial treatment of a 5-year old severely retarded child with a natural diet low in methionine. Am J Dis Child. 1967 Jan;113(1):95-7.
Carson NA, Carre IJ. Treatment of homocystinuria with pyridoxine. A preliminary study. Arch Dis Child. 1969 Jun;44(235):387-92.
De Franchis R, Sperandeo MP, Sebastio G, Andria G. Clinical aspects of cystathionine beta-synthase deficiency: how wide is the spectrum? The Italian Collaborative Study Group on Homocystinuria. Eur J Pediatr. 1998 Apr;157 Suppl 2:S67-70.
Gallagher, P. M.; Naughten, E.; Hanson, N. Q.; Schwichtenberg, K.; Bignell, M.; Yuan, M.; Ward, P.; Yap, S.; Whitehead, A. S.; Tsai, M. Y. : Characterization of mutations in the cystathionine beta-synthase gene in Irish patients with homocystinuria. Molec. Genet. Metab. 65: 298-302, 1998.
Grieco AJ. Homocystinuria: pathogenetic mechanisms. Am J Med Sci. 1977 Mar-Apr;273(2):120-32.
Grover VK, Malhotra SK, Kaushik S. Anaesthesia and homocystinuria. Anaesthesia. 1979 Oct;34(9):913-4.
Halpert M, BenEzra D. Surgery of the hereditary subluxated lens in children. Ophthalmology. 1996 Apr;103(4):681-6.
Harrison DA, Mullaney PB, Mesfer SA, Awad AH, Dhindsa H. Management of ophthalmic complications of homocystinuria. Ophthalmology. 1998 Oct;105(10):1886-90.
Juszko J, Kubalska J, Kanigowska K. [Ocular problems in children with homocystinuria]. Klin Oczna. 1994 Jun-Jul;96(6-7):212-5.
Kraus, J. P.; Janosik, M.; Kozich, V.; Mandell, R.; Shih, V.; Sperandeo, M. P.; Sebastio, G.; de Franchis, R.; Andria, G.; Kluijtmans, L. A. J.; Blom, H.; Boers, G. H. J.; Gordon, R. B.; Kamoun, P.; Tsai, M. Y.; Kruger, W. D.; Koch, H. G.; Ohura, T.; Gaustadnes, M. : Cystathionine beta-synthase mutations in homocystinuria. Hum. Mutat. 13: 362-375, 1999.
Michalski A, Leonard JV, Taylor DS. The eye and inherited metabolic disease: a review. J R Soc Med. 1988 May;81(5):286-90.
Richard JM, Friendly DS. Ocular findings in pediatric systemic disease. Pediatr Clin North Am. 1983 Dec;30(6):1123-44.
Simonelli F, De Crecchio G, Testa F, Nunziata G, Mazzeo S, Romano N, Cavaliere L, Rinaldi MM, Rinaldi E. Retinal degeneration associated with ectopia lentis. Ophthalmic Genet. 1999 Jun;20(2):121-6.
Streeten, B. W. : The nature of the ocular zonule. Trans. Am. Ophthal. Soc. 80: 823-854, 1982.

![]()
Suivez nous sur...
![]()
SNOF
10 rue Schweighaeuser
CS 40028
67080 STRASBOURG Cedex
Tél. 03 88 35 01 09
Fax. 03 88 25 51 90