Le site des ophtalmologistes de France
Espace Encyclopédie
Encyclopédie de la vue





Aniridie
Aniridie
Syndrome WAGR et syndrome de Gillespie
Aniridia
Aniridie
Définition
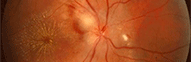
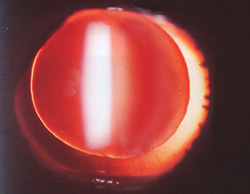
On définit l'aniridie comme une absence quasi totale et bilatérale d'iris. Cette anomalie est rapidement visible chez le petit enfant, ce qui entraîne une consultation précoce chez l'ophtalmologiste. Cette pathologie rare (1/40.000) se complique très souvent de glaucome, ce qui va nécessiter une surveillance très régulière de ces enfants.
Clinique
L'absence d'iris fait parfois penser que les pupilles sont très dilatées (mydriase). Il existe toujours une mince collerette d'iris et un important reflet orangé du fond d'oeil quand on l'éclaire, témoin de l'absence de myosis. On voit la totalité du cristallin et même la zonule de Zinn. Ces enfants sont souvent photophobes et ils ne sont pas très faciles à examiner à cause de cela.
Des anomalies de la cornée peuvent exister, comme des opacités, une sclérocornée ou des synéchies.
Il s'y associe souvent une dystrophie limbique qui va obérer les résultats d'une éventuelle greffe de cornée (kératoplastie transfixiante). Il est en effet possible que la cornée soit altérée et opaque dans le cadre d'une association avec un glaucome.
Une dysgénésie de l'angle irido-cornéen est fréquente (50 à 75% des cas) avec glaucome. Ces hypertonies intra-oculaires sont difficilement jugulées par des collyres, et il faut souvent intervenir chirurgicalement, en sachant que ces opérations sont parfois délicates à réaliser.
L'atteinte maculaire est quasi constante, avec un absence de macula ou une hypoplasie maculaire. Ceci entraîne un nystagmus et une basse vision (amblyopie).
Une cataracte ou des opacités cristalliniennes peuvent s'ajouter au tableau clinique.
Bilan familial
Il faut s'attacher à rechercher le type de transmission de cette anomalie, dans la famille de l'enfant.
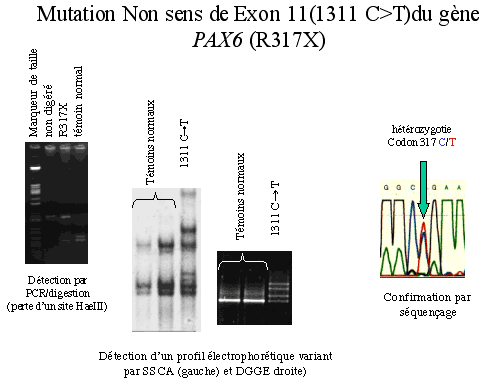
Il existe des familles dont plusieurs membres présentent une aniridie. Il s'agit alors d'une transmission génétique autosomique dominante par haplo-insuffisance du gène PAX6 qui est muté. Ce gène est capital dans l'embryologie de l'oeil.
Le problème survient quand l'enfant est le premier cas de la famille à présenter cette anomalie.
L'enfant peut être le premier d'une lignée génétique avec une transmission autosomique dominante dans le cadre d'une anomalie génétique (mutation de PAX6):
 Pr Patrick Calvas CHU Purpan Toulouse France |
La SSCA (Single Strand Conformation Analysis) et la DGGE (Denaturing Gradient Gel Electrophoresis) permettent de différencier des fragments d'ADN qui ont des aspects tridimensionnels dissemblables à cause d'une différence, même minime, de structure et donc de séquence. On pourra ainsi visualiser, en électrophrorèse, des migrations différentes. |
L'enfant peut parfois présenter une anomalie chromosomique, une délétion en 11p13. Cette délétion del11p s'accompagne du syndrome WAGR des anglo-saxons (Wilms tumor, Aniridia, Genito-urinary malformation, mental Retardation), décrit par Miler en 1964.
C'est donc l'association
- d'une tumeur de Wilms,
- d'une aniridie,
- d'une malformation génito-urinaire,
- et d'un retard mental.
Le syndrome WAGR est en rapport avec le gène WT1 qui est le gène suppresseur de la tumeur de Wilms, et qui est situé à proximité de PAX6. La mutation de ce gène entraînerait la production d'une protéine anormale WT1 qui annulerait l'action tumeur-suppressive du gène WT1 de l'allèle. Le gène WT1 suppresseur de tumeur a été transformé en un oncogène dominant négatif.
Ce gène WT1 contient 10 exons et code pour un facteur de transcription de développement des gonades et des reins.
Le risque d'un syndrome WAGR est de 30% pour un jeune patient présentant une aniridie non familiale.
Ces cas de délétion sont associés à un gonadoblastome ou un néphroblastome (tumeur de Wilms), qui sont des tumeurs graves mais curables. On s'attachera donc à rechercher ce type de tumeurs par une échographie abdominale tous les six mois, avec surveillance des organes génitaux externes du garçon. On décrit des malformations génito-urinaires (ambiguité sexuelle à ectopie testiculaire associée ou non à des anomalies urinaires).
Il est donc capital de demander un caryotype en haute résolution à la recherche de cette délétion parfois difficile à mettre en évidence, pour savoir dans quel groupe se situe l'enfant.
Analyse cytogénétique et caryotype:
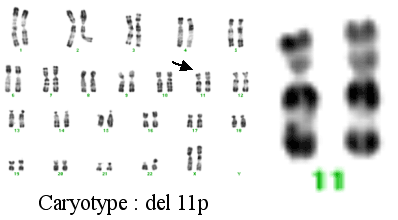
Caryotype avec délétion du chromosome 11 (del11)
Pr Patrick Calvas CHU Purpan Toulouse France
Hybridation in situ en fluorescence ou Fluorescence in situ hybridization (FISH) :
Pour évaluer le risque de tumeur de Wilms, on réalise une analyse cytogénétique par hybridation in situ en fluorescence (FISH), grâce à l'usage d'un panel de cosmides cernant le gène PAX6, le gène WT1 de prédisposition à la tumeur de Wilms, et des marqueurs associés, au niveau chromosomique 11p13. Quelques patients ont des délétions difficiles à mettre en évidence, seulement détectables au niveau 11p13 par FISH.
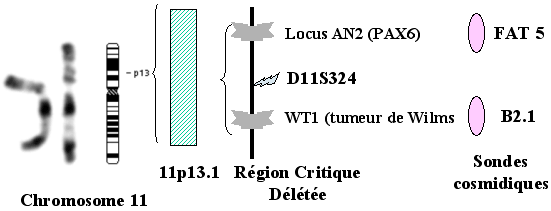
Syndrome WAGR
Pr Patrick Calvas CHU Purpan Toulouse France
La FISH (Fluorescence in Situ Hybridization ) consiste à repérer une région particulière du chromosome, grâce à une sonde oligonucléotidique complémentaire qui va s'hybrider avec l'ADN. Des antigènes sont couplés à certains des nucléotides et vont fixer des anticorps fluorescents. Grâce à un microscope UV on met en évidence, sous une lumière ultra violette, des spots de couleur.
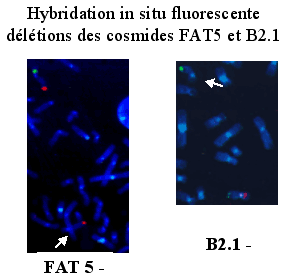
Hybridation in situ en fluorescence FISH
Pr Patrick Calvas CHU Purpan Toulouse France
Exemple d'étude FISH:
"FISH studies in patients with aniridia reveals >35% with chromosome abnormalities including five cryptic 3 PAX6 deletions. J.A. Crolla1, V. van Heyningen2. 1) Wessex Reg Genetics Lab, Salisbury District Hosp, Wiltshire, England; 2) MRC Human Genetics Unit, Western General Hospital Edinburgh EH4 2XU.
Seventy patients with aniridia (13 familial and 57 sporadic), referred for FISH and cytogenetic investigations, were studied initially using cosmids mapping to distal 11p13, (cen B2.1(WT1), D11S324, FAT5 {PAX6 exons 1-4}, and FO2121, tel). Finding a case with a deletion not involving FAT5 but including the marker FO2121, ~100kb telomeric, prompted additional studies on all cases reported as normal by FISH. A cosmid contig, mapping to a 180kb region 3` of PAX6, was used: (cen A1280 {PAX6 exons 5-13}, G0453, C1170, H1281, SRL11M20, SRL9A13, tel). Five of the 48 normal cases re-tested (7% of all cases) had small deletions ~50-430kb in size, all with a similar proximal breakpoint in cosmid A1280, indicated by diminished signal on one chromosome 11. One case with an ~180kb deletion was mosaic, with half the peripheral blood lymphocytes with the deletion and half with normal signals on both 11 homologues. Overall, 27 cases (38.6%) were chromosomally abnormal. Twelve (17%) had cytogenetically visible interstitial WAGR deletions involving 11p13, ten of which included WT1. Eleven (15.5%), including the five described above, had cryptic deletions only visible by FISH, two include WT1. The remaining four cases (5.5%) had chromosomal rearrangements: an unbalanced t(11;13) with a deletion of the WAGR region, and three (4%) balanced rearrangements with what appear to be position effect breakpoints 3 of PAX6; (a) an inv(11)(p13q13) with a breakpoint in cosmid H1281 (>75kb downstream of PAX6) (b) a t(7;11) with the 11p13 breakpoint ~30kb downstream of PAX6 and (c) a dir ins(12;11) with a breakpoint in cosmid C1170 (>40kb from PAX6). The rates and distribution of chromosome anomalies in familial (4/13, 30.8%) and sporadic cases (23/57, 40%) are similar. Whether the reduced A1280 signal indicates that the PAX6 gene is disrupted, remains to be assessed. If the 13 PAX6 exons are intact, these cases represent interstitial deletions resulting in position effects. As the five small deletions were not identified using cosmid FAT5, A1280 should be incorporated into diagnostic FISH procedures."
Une autre association a été décrite, c'est l'association d'une aniridie, d'une ataxie cérébelleuse et d'une déficience mentale, c'est le syndrome de Gillespie. Il n'y a pas de mutation de PAX6 et la transmission est autosomique récessive.
Conclusions
Il est important de surveiller très régulièrement les enfants présentant une aniridie, à cause du risque très élevé de glaucome.
La survenue de tumeurs en cas de délétion chromosomique doit aussi entraîner des contrôles fréquents.
Un bilan familial peut permettre de retrouver des marqueurs cliniques génétiques chez les ascendants ou les collatéraux.
Web et bibliographie
Association du syndrome WAGR (en anglais)
Groupe de discussion du syndrome WAGR (en anglais)
http://associationgeniris.free.fr
Axton, R.; Hanson, I.; Danes, S.; Sellar, G.; van Heyningen, V.; Prosser, J. : The incidence of PAX6 mutation in patients with simple aniridia: an evaluation of mutation detection in 12 cases. J. Med. Genet. 34: 279-286, 1997.
Baulmann DC, Ohlmann A, Flugel-Koch C, Goswami S, Cvekl A, Tamm ER. Pax6 heterozygous eyes show defects in chamber angle differentiation that are associated with a wide spectrum of other anterior eye segment abnormalities. Mech Dev. 2002 Oct;118(1-2):3-17.
Breslow, N. E.; Takashima, J. R.; Ritchey, M. L.; Strong, L. C.; Green, D. M. : Renal failure in the Denys-Drash and Wilms' tumor-aniridia syndromes. Cancer Res. 60: 4030-4032, 2000.
Crolla JA, Van Heyningen V. Frequent chromosome aberrations revealed by molecular cytogenetic studies in patients with aniridia. Am J Hum Genet. 2002 Nov;71(5):1138-49.
Gillespie, F. D. : Aniridia, cerebellar ataxia, and oligophrenia in siblings. Arch. Ophthal. 73: 338-341, 1965
Glaser, T.; Jepeal, L.; Edwards, J. G.; Young, S. R.; Favor, J.; Maas, R. L. : PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nature Genet. 7: 463-471, 1994
Glaser, T.; Ton, C. C. T.; Mueller, R.; Petzl-Erler, M. L.; Oliver, C.; Nevin, N. C.; Housman, D. E.; Maas, R. L. : Absence of PAX6 gene mutations in Gillespie syndrome (partial aniridia, cerebellar ataxia, and mental retardation). Genomics 19: 145-148, 1994.
Gronskov, K.; Olsen, J. H.; Sand, A.; Pedersen, W.; Carlsen, N.; Jylling, A. M. B.; Lyngbye, T.; Brondum-Nielsen, K.; Rosenberg, T. : Population-based risk estimates of Wilms tumor in sporadic aniridia: a comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum. Genet. 109: 11-18, 2001.
Gul, D.; Ogur, G.; Tunca, Y.; Ozcan, O. : Third case of WAGR syndrome with severe obesity and constitutional deletion of chromosome (11)(p12p14). (Letter) Am. J. Med. Genet. 107: 70-71, 2002.
Hanson, I.; Churchill, A.; Love, J.; Axton, R.; Moore, T.; Clarke, M.; Meire, F.; van Heyningen, V. : Missense mutations in the most ancient residues of the PAX6 paired domain underlie a spectrum of human congenital eye malformations. Hum. Molec. Genet. 8: 165-172, 1999.
Lorda-Sanchez I, Sanz R, Diaz-Guillen MA, Fernandez-Toral J, Heine-Suner D, Rodriguez De Alba M, Gonzalez-Gonzalez C, Trujillo MJ, Ramos C, Rodriguez De Cordoba S, Ayuso C. Aniridia as part of a WAGR syndrome in a girl whose brother presented hypospadias. Genet Couns. 2002;13(2):171-7.
Miller, R. W.; Fraumeni, J. F., Jr.; Manning, M. D. : Association of Wilms' tumor with aniridia, hemihypertrophy and other congenital malformations. New Eng. J. Med. 270: 922-927, 1964.
Muto R, Yamamori S, Ohashi H, Osawa M. Prediction by FISH analysis of the occurrence of Wilms tumor in aniridia patients. Am J Med Genet. 2002 Apr 1;108(4):285-9.
Sale MM, Craig JE, Charlesworth JC, FitzGerald LM, Hanson IM, Dickinson JL, Matthews SJ, Heyningen Vv V, Fingert JH, Mackey DA. Broad phenotypic variability in a single pedigree with a novel 1410delC mutation in the PST domain of the PAX6 gene. Hum Mutat. 2002 Oct;20(4):322.
Sisodiya, S. M.; Free, S. L.; Williamson, K. A.; Mitchell, T. N.; Willis, C.; Stevens, J. M.; Kendall, B. E.; Shorvon, S. D.; Hanson, I. M.; Moore, A. T.; van Heyningen, V. : PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nature Genet. 28: 214-216, 2001.
van Heyningen V, Williamson KA. PAX6 in sensory development. Hum Mol Genet. 2002 May 15;11(10):1161-7. Review.

![]()
Suivez nous sur...
![]()
SNOF
10 rue Schweighaeuser
CS 40028
67080 STRASBOURG Cedex
Tél. 03 88 35 01 09
Fax. 03 88 25 51 90