Le site des ophtalmologistes de France
Espace Encyclopédie
Encyclopédie de la vue

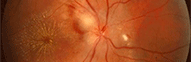




Atrophie Optique Dominante Labo CHU Angers
Présentation de l'équipe du CHU d'Angers
Le laboratoire de Biochimie du CHU d’Angers (Pr Pascal Reynier et Dr Patrizia Amati-Bonneau), en étroite collaboration avec le Service de Génétique Médicale (Pr Dominique Bonneau) propose une étude menée sur 2 ans pour préciser les caractéristiques cliniques et génétiques de l’atrophie optique dominante (AOD).
L’AOD est une pathologie du nerf optique conduisant à une perte progressive de l’acuité visuelle, à une perturbation de la vision des couleurs et à une atrophie du nerf optique visible au fond d’œil. Dans cette affection, il existe une très grande variabilité clinique inter et intra familiale.
Le gène OPA1, récemment identifié sur le bras long du chromosome 3, est le gène responsable de plus de 95% des cas d’atrophie optique dominantes.
Les objectifs de notre étude sont :
1) Obtenir les informations les plus précises possible sur le degré de gravité de la maladie en relation avec les mutations du gène OPA1.
2) Collecter des prélèvements de sujets et de familles pour lesquels aucune mutation du gène OPA1 n’a été mise en évidence.
Les patients seront recrutés à partir des différents Services de Génétique et d’Ophtalmologie qui collaborent actuellement avec notre unité. Le gène OPA1 sera étudié par les méthodes classiques de la génétique moléculaire. Il est également envisagé de réaliser un site internet accessible à tout laboratoire travaillant sur OPA1 pour essayer de coordonner au moins au niveau européen les recherche sur l’AOD.
Dr Patrizia Amati-Bonneau

Diagnostic Moléculaire de l'Atrophie Optique Dominante (AOD)
L’AOD et la neuropathie optique héréditaire de Leber sont les deux formes les plus fréquentes d’atrophie optique d’origine génétique.
L’existence de formes familiales d’atrophie optique a été montrée par Batten en 1896 et par Snell en 1897. Cependant, c’est l'ophtalmologue danois Kjer qui en 1959 a établi la possible transmission autosomique dominante de la maladie en rapportant une étude sur 19 familles (4).
L’AOD, aussi dénommée atrophie optique de Kjer, a une prévalence globale de 1/50 000 et de 1/10000 au Danemark (2).
Phénotype clinique
L’AOD se caractérise cliniquement par
- une baisse progressive de l’acuité visuelle,
- une atrophie du nerf optique,
- un scotome centro-cæcal et
- une dyschromatopsie dans l’axe bleu-jaune.
L’AOD est souvent diagnostiquée dans l’enfance, mais elle peut également débuter dès la première année de vie et être suspectée devant un nystagmus.
L’atrophie optique, bilatérale et symétrique, est mise en évidence par l’examen du fond de l’œil sous forme d’une pâleur temporale de la papille.
Ces lésions sont dues à la perte des fibres nerveuses venant de la macula. Il n’y a pas d’excavation au niveau du nerf optique, ce qui différencie l’AOD d’une atrophie optique due à un glaucome.
La réduction du nombre des fibres du nerf optique est aussi responsable du scotome centro-cæcal.
La baisse de l’acuité visuelle est variable d’un individu à l’autre. En général, elle reste modérée (de 6/10 à 2/10), mais elle peut quelquefois conduire à la cécité. Certains individus porteurs du gène muté peuvent conserver une acuité visuelle normale.
Dans l’AOD, l’anomalie de la vision des couleurs se fait dans l’axe bleu-jaune (tritanopie) à la différence des autres types d’atrophies optiques (comme l’atrophie optique de Leber) où l’axe de confusion est rouge-vert.
L’AOD a une pénétrance incomplète et une expressivité intra et inter-familiale variable.
Sur le plan histopathologique, l’AOD se caractérise par une dégénérescence de cellules ganglionnaires de la rétine et une perte de la myéline et du tissu nerveux au niveau du nerf optique, comme celle que l’on observe dans la neuropathie optique héréditaire de Leber. Cette dernière affection constitue le principal diagnostic différentiel de l’OAD ; elle se caractérise par une baisse plus brutale de l’acuité visuelle et par une transmission maternelle.
Génétique moléculaire
Le tableau ci-dessous résume les quatre loci qui ont été identifiés à ce jour pour les atrophies optiques, deux d’entre eux, OPA1 et OPA4, concernent les formes dominantes de la maladie.
|
Symbole |
Transmission |
MIM |
Locus |
Gène |
|
OPA1 |
AD |
165500 |
3q28-q29 |
OPA1 |
|
OPA2 |
RLX |
60593 |
Xp11 |
Non identifié |
|
OPA3 |
AR |
606580 |
19q13 |
MGA |
|
OPA4 |
AD |
605293 |
18q12 |
Non identifié |
Le gène OPA1 a été localisé sur l’extrémité terminale du bras long du chromosome 3 par Eiberg en 1994 par une étude de liaison sur 3 familles danoises (5). Cette localisation à été confirmée sur des familles d’origine cubaine (6), française (7), britannique (8, 9) et nord-américaine (10). Le locus OPA1, en 3q28-29, est le locus majeur responsable de l’affection, cependant l’hétérogénéité génétique de l’AOD a été démontrée par la mise en évidence d’un deuxième locus en 18q12.2-12.3 (ref 11). A ce jour, la liaison à ce locus n’a été reconnue que dans une seule famille.
Le gène OPA1 a été récemment identifié par une équipe française (1) et par une équipe britannique (12). Il s’agit d’un gène codant pour une dynamine/GTPase impliquée dans la maintenance et la division desmitochondries. Le gène OPA1 a une homologie de séquence de 33% avec le gène Msp1 de la levure Schizosaccharomyces pombe et de 31% avec le gène orthologue du Saccharomyces Cerevisiae Mgm1p (maintenance génome mitochondrial). Les trois protéines Msp1, Mgm1p et OPA1 sont constituées d’un domaine GTPase, d’un domaine central de dyamine très conservé, et d’une partie N-terminale nécessaire pour l’adressage dans la mitochondrie (13, 14).
Le gène humain OPA1 s’étend sur environ 90 kb et possède 31 exons dont 30 sont codants.
Le cDNA a une taille de 5821 nucléotides (dont 2878 nucléotides codants) et la protéine est formée de 960 acides aminés. OPA1 est exprimé dans tous les tissus mais surtout dans la rétine (1, 12). Les études d’hybridation in situ chez le rat et la souris montrent qu’OPA1 a une expression très forte au niveau des cellules ganglionnaires de la rétine.
A ce jour, 62 mutations d’OPA1 ont été identifiées chez 201 malades (15). La grande majorité (90%) de ces mutations sont situées entre l’exon 8 et 28 et intéressent surtout le domaine GTPase (54%) ; la moitié d’entre-elles sont des mutations tronquantes. La mutation 2708delTTAG dans l’exon 27 touche 12.5% de toutes les familles analysées par les différentes équipes (15). Cette délétion correspond à unpoint chaud de mutation et non à une mutation ancestrale puisqu’elle ne survient pas sur le même fond génétique dans les différentes familles étudiées (16). En revanche, un effet fondateur a été retrouvé au Danemark pour la mutation c.2826delT dans l’exon 28 (ref 17).
Résultats obtenus dans notre laboratoire
Depuis la découverte du gène OPA1, nous avons collecté l’ADN de 160 sujets atteints de neuropathie optique. Il s’agit de cas familiaux ou sporadiques provenant de différents services de Génétique et d’Ophtalmologie de France. Le diagnostic moléculaire a été réalisé par séquençage direct des 30 exons codants d’OPA1 pour les patients atteints spécifiquement d’AOD. Nous avons mis en évidence 14 nouvelles mutations d’OPA1 (manuscrit en préparation); l’une d’entre-elles est responsable d’une forme congénitale d’atrophie optique.
Actuellement, notre laboratoire est le seul à réaliser en France le diagnostic moléculaire de l’AOD.
Un projet de réalisation d’un site internet dédié à OPA1 est en cours d’elaboration. Il s’agit de créer un site accessible à tout laboratoire travaillant sur OPA1 pour essayer de coordonner (au moins au niveau européen) les laboratoires faisant le diagnostic de l’AOD et pour tenter de d’élargir la série de sujets atteints pour de meilleures corrélations génotype-phénotype. Ce site se propose également de tenir à jour la bibliographie sur les atrophies optiques. Cette collaboration pourrait permettre en outre de collecter un plus grand nombre de famille OPA1 négatives en vue de la localisation d’autres gènes impliqués dans l’atrophie optique.
Bibliographie
Delettre C, Lenaers G, Griffoin J-M et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 2000; 26: 207-210.1.
Votruba M, Moore T, Bhattacharya S. Clinical features, molecular genetics, and pathophysiology of dominant optic atrophy. J Med Genet, october 1998; 35: 793-800.
Man PY, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. J Med Genet 2002 ; 39 : 162-169.
Kjer P . Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol Scand 1959; 37 [Suppl 54]:1-146.
Eiberg H, Kjer B, Kjer P and Rosemberg T. Dominant optic atrophy (OPA1) mapped to chromosome 3q region. Linkage analysis. Hum Mol Genet 1994; 3: 3977-3980.
Lunkes A, Hartung U, Magarino C, Rodriguez M, Palmero A, Rodriguez L, Heredero
L, Weissenbach J, Weber J, Auburger G. Refinement of the OPA1 gene locus on chromosome 3q28-q29 to a region of 2-8 cM, in one Cuban pedigree with autosomal dominant optic atrophy type Kjer. Am J Hum Genet 1995; 57: 968-970.
Bonneau D, Souied E, Gerber S et al. No evidence of genetic heterogeneity in dominant optic atrophy. J Clin Genet ,1995, 32 :951-953.
Votruba M, Moore AT, Bhattacharya SS. Genetic refinement of dominant optic atrophy (OPA1) locus to within a 2 cM interval of chromosome 3q. J Med Genet 1997; 34:117-121.
Johnston RL, Seller MJ, Behnam JT, Burdon MA, Spalton DJ. Dominant optic atrophy. Refining the clinical diagnostic criteria in light of genetic linkage studies. Ophthalmology 1999; 106: 123-128.
Brown J Jr, Fingert JH, Taylor CM, Lake M, Sheffield VC, Stone EM. Clinical and genetic analysis of a family affected with dominant optic atrophy (OPA1). Arch Ophthalmol 1997; 115: 95-99.
Kerrison JB, Arnould VJ, Ferraz sallum JM, et al. Genetic heterogeneity of dominant optic atrophy, Kyjer type: identification of a second locus on chromosome 18q22-23. Arch Ophthalmol 1999; 117: 805-810.
Alexander C, Votruba M, Pesch UE et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 2000; 26: 211-215.
Otsuga D, Keegan BR, Brisch E et al. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J Cell Biol 1998; 143: 333-349.
Pelloquin L, Belenguer P, Menon Y et al. Identification of a fission yeast dynamin-related protein involved in mitochondria DNA maintenance. Biochem Biophys Res Commun 1998; 251: 720-726.
Delettre C, Lenaers G, Pelloquin L, Belenguer P, Hamel CP. OPA1 (Kjer Type) Dominant Optic Atrophy: A Novel Mitochondrial Disease. Mol Genet Metab 2002; 75: 97-107.
Toomes C, Marchbank NJ, Mackey DA et al. Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy. Hum Mol Genet 2001, 10: 1369-1378.
Thiselton DL, Alexander C, Morris A et al. A frameshift mutation in exon 28 of the OPA1 gene explains the high prevalence of dominant optic atrophy in the Danish population: evidence for a founder effect. Hum Genet 2001; 109: 498-502.
Publications du laboratoire
Reynier P, Malthièry Y, Lestienne P. Mitochondrial DNA analysis in « Mitochondrial diseases » by P Lestienne (Ed.). Springer-Verlag 1999, chapter 27, 379-387.
Reynier P, Penisson-Besnier I, Moreau C, Savagner F, Vielle B, Emile J, Dubas F, Malthièry Y. mtDNA haplogroup J: a contributing factor of optic neuritis. European Journal of Human Genetics 1999, 7: 404-406.
Penisson-Besnier I, Moreau C, Jacques C, Roger JC, Dubas F, Reynier P. Sclérose en plaques et mutations de l’ADN mitochondrial associées à la maladie de Leber. Revue Neurologique (Paris) 2001, 157 : 537-541.
Reynier P, Savagner F, Malthièry Y. Analyse de l’ADNmt humain in “Les Maladies Mitochondriales” Editeurs scientifiques: Cohen GN et Lestienne P, Annales de l’Institut Pasteur / actualités, Elsevier 2001 pages 55-70.
Funalot B, Reynier P, Vighetto A, Ranoux D, Bonnefont JP, Godinot C, Malthièry Y, Mas JL. A Leigh-like mitochondrial encephalopathy complicating Leber’s Hereditary Optic Neuropathy.Annales of Neurology, 2002, 52 : 374-377.
Baris O, Delettre C, Amati-Bonneau P, Surget MO, Charlin JF, Catier A, Dollfus H, Jonveaux P Ayuso C, Maumenee I, Lorenz B, Mohammed S, Tourmen Y, Bonneau D, Malthièry Y, Hamel C, Reynier P . 14 Novel opa1 mutations in dominant optic atrophy and de novo mutations in isolated cases of optic atrophy. Hum Mutation In revision
JD Gicquel, P Vabres, D Bonneau, M Mercié, Latifa Handiri, Paul Dighiero. Retinal Angioma in a patient with Cowden Disease. American Journal of Ophthalmology, 2003, 135, 400-402.
Vabres P, Bonneau D, Larregue M. Absence of lisch nodules in sporadic neurofibromatosis type 1 may reflectsomatic mosaicism. Archives of Dermatology 2002; 138: 839-840.
T Hearn, G L Renforth, C Spalluto, N A Hanley, K Piper, S Brickwood, C White, V Connolly, J F N Taylor, I Russell-Eggitt, D Bonneau, M Walker, D I Wilson Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. Nature Genetics 2002, 31, 79-83
D Y Nishimura, C C Searby, R Carmi, K Elbedour, L Van Maldergem, A B Fulton, B L. Lam, B R Powell, R E Swiderski, K E Bugge, N B Haider, A E Kwitek-Black, L Ying, D M Duhl, S M Gorman, E Heon, A Iannaccone, D Bonneau, L G Biesecker, S G Jacobson, E M Stone, V C Sheffield. Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome (BBS2). Human Molecular Genetics 2001, 15, 865-874.
Crisponi L, Deiana M, Loi A, Chiappe F, Uda M, Amati P, Bisceglia P, Zelante L, Nagaraja R, Porcu S, Ristaldi MS, Marzella R, Rocchi M, Nicolino M, Lienhardt-Roussie A, Nivelon A, Schlessinger D, Gasparini P, Bonneau D, Cao A, Pilia G. The Forkhead Transcription Factor FOXL2 Gene is Mutated in both Type II and Infertility-associated Type I Blepharophimosis/Ptosis/Epicanthus Inversus Syndrome (BPES). Nature Genetics, 2001, 27, 159-166
Bisceglia L, d'Ambrosio L, Piemontese MR, Carella M, Amati P, Bonneau D, Pilia G, Gasparini G, Zelante L. Cellular retinol binding protein 1 (RBP1): a frequent polymorphism, refined map position and exclusion as the blepharophimosis ptosis epicanthus inversus syndrome gene. Molecular Cell Probes 1998, 12, 255-258.
MR Piemontese, E Memeo, M Carella, P Amati, D Bonneau, G Pilia, A Cao, H Drabkin, R Gemmill, J Rommens, L Zelante, P Gasparini, L Bisceglia. A YAC contig spanning the blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) and propionic acidemia (PCCB) loci. Europan Journal of Human Genetic 1997, 5, 171-174.
P Amati, P Gasparini, J Zlotogora, JC Chomel, J Kaplan, A Kitzis, D Bonneau. A gene for premature ovarian failure and eyelid malformation maps to chromosome 3q22-q23. (Letter) American Journal of Human Genetics 1996, 58, 1089-1092.
S Gerber, D Larget-Piet, JM Rozet, D Bonneau, M Mathieu, V der Kaloustian, A Munnich, J Kaplan. Evidence for a fourth locus in Usher syndrome type I. Journal of Medical Genetics 1996, 33, 77-79.
C Beaumont, P Leneuve, I Devaux, M Berthier, MN Loizeau, H Grandchamp, D Bonneau. Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinemia and cataract. Nature Genetics 1995, 11, 444-446.
D Bonneau, E Souied, S Gerber, JM Rozet, E D'Haens, H Journel, G Plessis, J Weissenbach, A Munnich, J Kaplan. No evidence of genetic heterogeneity in dominant optic atrophy. Journal of Medical Genetics 1995, 32, 951-953.
D Bonneau, I Winter-Fuseau, MN Loiseau, P Amati, M Berthier, D Oriot, C Beaumont. Bilateral cataract and high serum ferritin: a new dominant disorder? Journal of Medical Genetics 1995, 32, 778-779.
A Camuzat, H Dolfus, JM Rozet, S Gerber, D Bonneau, M Bonnemaison, ML Briard, JL Dufier, I Ghazi, C Leowski, J Weissenbach, J Frézal, A Munnich, J Kaplan. A gene for Leber's congenital amaurosis maps to chromosome 17p. Human Molecular Genetics 1995, 4, 1447-1452.
P Amati, JC Chomel, A Nivelon-Chevalier, S Gingelkrantz, A Kitzis, J Kaplan, D Bonneau. A gene for blepharophimosis, ptosis, epicanthus inversus maps to chromosome 3q23. Human Genetics 1995, 96, 213-215.
S Gerber, JM Rozet, D Bonneau, M Goldschild, E Souied, J Frézal, A Munnich, J Kaplan. Exclusion of the cone-specific alpha-subunit of the transducin gene in Stargardt's disease. Human Genetics 1995, 95, 382-384.
S Gerber, JM Rozet, D Bonneau, E Souied, A Camuzat, JL Dufier, A Munnich, J Kaplan. A gene for fundus flavimaculatus with macular dystrophy of late onset maps on chromosome 3q. American Journal of Human Genetics 1995, 56, 396-399.
E Souied, S Gerber, JM Rozet, D Bonneau, JL Dufier, I Ghazi, N Philip, G Soubrane, G Coscas, A Munnich, J Kaplan. Five novel mutations of the rhodopsin gene in autosomal dominant retinitis pigmentosa. Human Molecular Genetics 1994, 3, 1433-1434.
D Larget-Piet, S Gerber, D Bonneau, JM Rozet, I Gazzi, JL Dufier, A David, P Bitoun, A Munnich, J Kaplan. Genetic heterogeneity of Usher Syndrome type I in french families. Genomics 1994, 21, 138-143.
D Bonneau, F Raymond, F Patte, C Kremer, JM Klossek, J Kaplan. Usher syndrome associated with bronchiectasis in two brothers. Journal of Medical Genetics 1993, 30, 253-254.
J Kaplan, S Gerber, D Bonneau, JM Rozet, ML Briard, H Dolfus, JL Dufier, J Frézal, A Munnich. Usher syndrome type 1 maps to chromosome 14 q. Genomics 1992, 14, 979-987.
J Kaplan, A Pelet, C Martin, O Delrieu, S Aymé, D Bonneau, ML Briard, A Hanauer, L Larget-Piet, P Lefrançois, A Michel-Awad, H Plauchu, JL Dufier, J Frézal, A Munnich. Phenotype-genotype correlations in retinitis pigmentosa. Journal of Medical Genetics 1992, 29, 615-623.
D Bonneau, J Kaplan, G Girard, JL Dufier. Autosomal recessive inheritance of senile retinitis pigmentosa. A report of one family with consanguinity. Clinical Genetics 1992, 42, 199-200.
J Kaplan, D Bonneau, J Frézal, A Munnich, JL Dufier. Clinical and genetic heterogeneity in retinitis pigmentosa. Human Genetics 1990, 85, 635-642.
D Bonneau, J Kaplan, M Berthier, J Seger, JL Dufier, J Frézal. Evidence against linkage of the gene for Usher's syndrome type 1 with group specific component (GC) on chromosome 4. Annales de Génétique 1990, 33, 103-104.
J Kaplan, G Guasconi, D Bonneau, J Melki, M L Briard, A Munnich, JL Dufier, J Frézal. Usher syndrome type 1 is not linked to D1S81 (pTHH 33): evidence for genetic heterogeneity. Annales de Génétique 1990, 33, 105-108.

![]()
Suivez nous sur...
![]()
SNOF
10 rue Schweighaeuser
CS 40028
67080 STRASBOURG Cedex
Tél. 03 88 35 01 09
Fax. 03 88 25 51 90