Le site des ophtalmologistes de France
Espace Encyclopédie
Encyclopédie de la vue

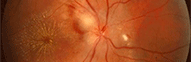




Maladie de Refsum
Maladie de Refsum
Heredopathia atactica polyneuritiformis
Refsum disease
Call to Research project on Refsum disease <=>Projet de recherche clinique sur la maladie de Refsum adulte

Sigvald Refsum
By courtesy of Refsum family
La réalisation de cette page n'aurait pas été possible sans l'aide du Professeur Patrick Aubourg (Hôpital Saint-Vincent de Paul, Paris France). Nous le remercions beaucoup.
Définition
Il s'agit d'une maladie héréditaire du métabolisme, autosomique récessive, décrite en 1946 par le Norvégien Sigvald Bernhard Refsum (1907-1991).
Cette maladie est due à un mauvais fonctionnement d'une enzyme localisée dans les péroxysomes qui sont des organites intra-cellulaires dont les fonctions sont multiples:
- la dégradation de l'H2O2 produite par les oxydases (rôle antioxydant des péroxysomes),
- la biosynthèse de phospholipides à liaison éther (plasmalogènes),
- et l'oxydation des acides gras à longue chaîne (AGL), à très longue chaîne (AGTLC), et d'acides gras branchés, dont l'acide phytanique.
Les péroxysomes contiennent plus de 60 enzymes et sont particulièrement abondants dans le foie et le rein, mais aussi le système nerveux central et périphérique, ainsi que la rétine.
L'acide phytanique est un acide gras issu des aliments. Il provient principalement de la dégradation de la chlorophylle. On le trouve dans les végétaux et les produits des herbivores (viande, lait, beurre...).
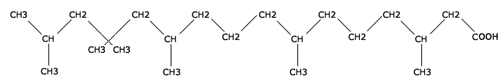
Acide phytanique
L'acide phytanique doit être régulièrement dégradé par ß-oxydation, comme tous les acides gras, et cette dégradation ne se fait que dans le péroxysome (la plupart des acides gras sont dégradés dans la mitochondrie).
L’acide phytanique doit d’abord être activé en ester de CoenzymeA (c'est le phytanoyl CoA), mais celui-ci ne peut être ß-oxydé directement. Le phytanoyl CoA doit être hydroxylé en 2-hydroxy-phytanoyl-CoA par une enzyme appellée phytanoyl-CoA hydroxylase (PAHX ou PHYH). C’est cette enzyme qui est déficiente dans la maladie de Refsum adulte. Le 2-hydroxy-phytanoyl-CoA est ensuite converti en acide pristanique qui peut ensuite être ß-oxydé dans le péroxysome (Fig 1).
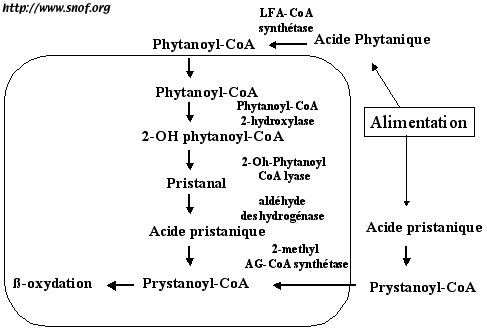
Fig. 1: Métabolisme de l'acide phytanique
Le déficit d’activité de la PAHX provoque une accumulation progressive d'acide phytanique (acide 3,7,11,15-tétraméthylhexadécanoïque) dans tous les organes de l'organisme, mais provoque des lésions essentiellement dans la rétine, le système nerveux central (SNC) et les nerfs périphériques.
Il existe d’autres maladies péroxysomales comportant une accumulation d’acide phytanique, mais celles-ci s’accompagnent toujours d’une accumulation d’un autre acide gras, l’acide pristanique, et souvent d’autres métabolites :
1°) le déficit en alpha-méthylacyl-CoA racémase (qui convertit les stéréoisomers R du phytanoyl-CoA en stéréoisomers S) (Fig.2) provoque une accumulation d’acide phytanique et pristanique, et une neuropathie sensitivo-motrice avec ou sans rétinite pigmentaire (comme dans le Refsum adulte). McLean BN, Allen J, Ferdinandusse S, Wanders RJ. A new defect of peroxisomal function involving pristanic acid: a case report.J Neurol Neurosurg Psychiatry 2002 Mar;72(3):396-9
2°) le déficit de la deuxième enzyme (protéine D-bifonctionnelle) de la ß-oxydation péroxysomale des AGTLC provoque une accumulation d’acide phytanique et pristanique et des intermédiaires de la synthèse des acides biliaires. Ce déficit entraîne une atteinte neurologique grave dès la naissance ou les premiers mois de vie conduisant au décès avant l’âge de 2 ans.
3°) les maladies génétiques de la biogénèse du péroxysomes entraînent une atteinte neurologique grave dès la naissance, mais qui peut se manifester parfois seulement à l’adolescence ou à l’âge adulte. Les formes les moins sévères de ces maladies ont été décrites dans le passé comme maladie de « Refsum infantile », parce que le premier métabolite que l’on ait trouvé, s’accumulant anormalement, a été l’acide phytanique. Ces maladies sont cliniquement, biochimiquement, et génétiquement totalement différentes de la maladie de Refsum adulte.
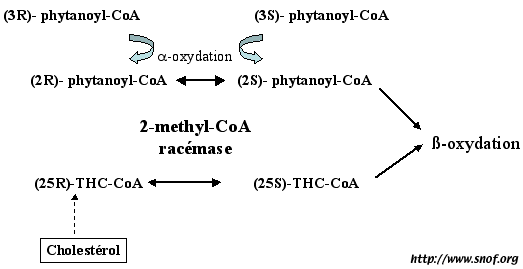
Fig. 2: Action de l'alpha-méthylacyl-CoA racémase
Génétique
La grande majorité des maladies de Refsum adulte sont dues à des mutations du gène PAHX localisé en 10pter-p11.2, et ces mutations altèrent l’activité enzymatique de l’enzyme.
Une minorité des patients atteints de maladie de Refsum n’ont pas de mutation du gène PAHX. Ils ont par contre une mutation d’une protéine (pex 7) impliquée dans la biogenèse des péroxysomes. Cette protéine est un récepteur qui permet de cibler à la surface des péroxysomes les enzymes qui ont un motif d’adressage de type PTS2 (Arg/Lys-Leu-X5-Gln/His-Leu). La phytanoyl-CoA hydroxylase a précisément ce motif d’adressage PTS2. En cas de mutation de son récepteur (la péroxine 7) dans le cytosol de la cellule, l’enzyme PAHX ne peut être ciblée dans le péroxysome (Fig. 3). D’autres mutations du gène PEX 7 donnent une maladie cliniquement totalement différente (chrondrodysplasie rhyzomélique).
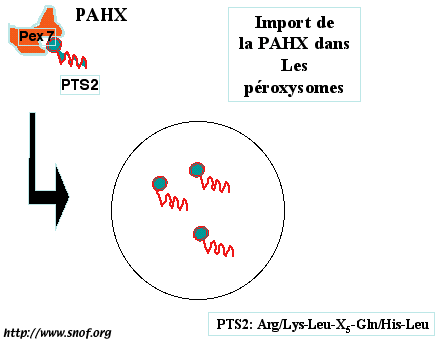
Fig. 3: Le motif d'adressage PTS2
Clinique
Les signes cliniques de cette maladie commencent entre 5 et 20 ans. Il existe souvent une grande variabilité dans l’intensité et la date d’apparition de chaque symptôme, même parmi les patients d’une même famille.
On décrit différents groupes de signes cliniques:
=> une rétinopathie pigmentaire,
=> une ataxie cérebelleuse,
=> une polyneuropathie distale motrice symétrique,
=> et parfois
- un syndrome pyramidal
- une dysarthrie
- une anosmie
- une surdité
- et une atteinte cardiaque avec troubles de la conduction.
On peut aussi parfois observer une ichtyose cutanée et une dysplasie modérée des épiphysaires.
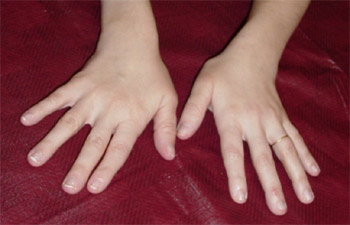
Coup de vent cubital
Latéro-flexion de la main vers le bord cubital
Maladie de Refsum
Le dosage plasmatique de l’acide phytanique permet de porter le diagnostic. Il doit être confirmé par la mesure de l’oxydation de l’acide phytanique dans les fibroblastes et la recherche de la mutation du gène PAHX. Il est important d’éliminer d’autres maladies péroxysomales qui peuvent donner parfois un phénotype clinique voisin (déficit en racémase, anomalies de la biogénèse des péroxysomes).
Traitements
Pour l'instant il n'y a pas de traitement de l'anomalie génétique.
On a proposé de combattre la thésaurismose d'acide phytanique par un régime alimentaire sans produits végétaux et sans aliment contenant de l'acide phytanique. Ce régime est très difficile à suivre et n’a, à lui seul, que peu d’effets sur la maladie. Par contre, sa combinaison avec des plasmaphérèses régulières permet de diminuer l'importance de l'ichtyose, et stabiliser la neuropathie et l’atteinte cardiaque. L’effet de ce traitement sur l’atteinte oculaire et la surdité est moins certaine. Il est probable cependant que plus ce traitement est commencé tôt, plus il a de chance d’être efficace.
Régime diététique pour la maladie de Refsum adulte
Schématiquement :
- - pas de viandes rouges : blanc de volaille
- - poissons maigres
- - 2 œufs par semaine pas plus
- - fruits de mer autorisés
- - pattes, riz
- - légumes uniquement qui poussent sous la terre
- - fruits épluchés et sans pépins
- - laitage : 0% matière grasse
- - margarine type « Fruit d’Or ».
Alimentation à environ 70 g de protides et 2700 calories, limitée en acide phytanique.
Exemple de répartition au cours de la journée en fonction de vos goûts et habitudes alimentaires avec un apport calorique suffisant pour éviter tout catabolisme entraînant une lipolyse.
Petit déjeuner :
- 1 café sucré
- 1 petit pain + confitures ou miel
- 1 ou 2 verres de jus de fruits : orange, pamplemousse, citron + sucre
Déjeuner :
- Crudités en salade assaisonnées à l‘huile de tournesol (1 cuillère à soupe)
- Viande ou poisson (voir liste)
- Pommes de terre (ou pâtes ou riz) + 1 cuillère à soupe d’huile de tournesol
- 1 petit pain (ou _ de baguette)
- Fruits au sirop
- 1 verre de jus de fruits sucré
Goûter :
- 1 petit pain
- 1 compote + 1 fromage blanc à 0 % de matières grasses ou 1 yaourt à 0 % de matières grasses + sucre
- 1 verre de jus de fruit sucré ou 1 café bien sucré
Soir :
- Repas identique à celui du déjeuner avec des légumes en plus.
Dans la soirée :
- 1 bouillie : 1 à 2 cuillerée à soupe de Maïzena à faire cuire dans _ de litre de lait écrémé ; lorsque la Maïzena est cuite, ajouter du sucre et un parfum : caramel, vanille ou extrait de fruits.
Les aliments strictement interdits sont principalement les aliments contenant des graisses telles que :
- - Graisses du lait et des laitages : fromage, fromage blanc, yaourts au lait entier ou desserts gélifiés au lait entier ou dessert à base de crème fraîche.
- - Graisses contenues dans les fruits secs ou oléagineux : olives, noix, noisettes, amandes, cacahuètes.....
- - Chocolat.
- - Viande des ruminants : bœuf, mouton, chevreuil....
- - Abats, lard.
- - Poissons gras : poissons bleus. Exemples de poissons maigres : bar, colin, cabillaud, carrelet, dorade, églefin, limande, sole, merlan, raie ; coquillages (coques, moules) ; seiche ; crabe ; crevettes.
L’origine de l’acide phytanique et de son précurseur le phytol serait alimentaire à plus de 90 %.
Un régime standard (il est très difficile de l’affirmer dans la mesure où la teneur en ces 2 constituants n’a été que très peu étudiée) apporterait en moyenne 60 mg/jour d’acide phytanique.
Le phytol libre est bien absorbé par l’intestin (60 à 90 %de la dose ingérée) dont 10 à 20 % sont transformés en acide phytanique.
Par contre, le phytol lié à la chrorophylle contenue dans les légumes verts est en grande partie éliminée dans les selles.
Un régime standard apporterait en moyenne 2 mg/jour de phytol libre.
=> Lors de la découverte de la maladie, alors que les taux sériques d’acide phytanique sont maximaux (avec des troubles moteurs graves) et selon la compliance du malade, un régime très strict peut être instauré ne contenant pas d’acide phytanique et/ou de phytol (1ère étape).
Le régime est un régime liquide, difficile à suivre sur de très longues périodes car peu varié et pouvant être plus ou moins bien toléré entraînant anorexie, vomissements ou diarrhée. Mais il a pour but :
- De faire diminuer rapidement l’accumulation d’acide phytanique.
- D’apporter suffisamment de calories pour éviter tout catabolisme entraînant une lipolyse.
- D’éviter les périodes de jeûne puisqu’il sera pris à intervalles réguliers au cours de la journée (en 4 ou 5 prises).
Il se compose de la façon suivante :
- 150 g de poudre de lait écrémé.
- 30 g Alburone ou Hyperprotidine.
- 200 g Dextrine Maltose ou Caloreen } composition pour 2 litres
- 20 g Huile de maïs.
Et sera donné à raison de 2 ou 2,5 litres par jour
(voir les supplémentations vitaminiques, minérales et oligo-éléments).
=> La 2ème étape du régime est de réintroduire l’acide phytanique (et le phytol libre ou lié à la chlorophylle), mais de façon à obtenir un régime apportant < 10 mg d’acide phytanique par jour.
Pour ce faire, différentes listes d’aliments ont été faites.
Liste d’aliments permis, à volonté:
- Lait écrémé, yaourts maigres
- Poulet, dinde, lapin – Poissons maigres (voir liste)
- Œufs
- Fruits en boîte (pas les fruits verts)
- Orange fraîche et pamplemousse frais, citron
- Betteraves, carottes, champignons, oignons, navets, radis
- Lentilles
- Huile de tournesol
- Thé, boissons gazeuses, bière, pulpes de fruits, café, eau, coca
- Confitures, gelées, sirops, mélasse, miel
- Epices, moutarde
Liste d’aliments interdits (graisses des laitages et viandes des ruminants):
- Lait entier, lait concentré
- Yaourts, fromage blanc, crème
- Bœuf, agneau, abats, poissons gras
- Maïs, pop-corn
- Pommes, bananes, groseilles, raisin, rhubarbe, fruits secs, noix
- Tous les légumes verts
- Huile végétale, huile d’arachide, beurre, margarine, lard, saindoux
- Chocolat, entremets
- Fines herbes fraîches
Liste d’aliments contrôlés:
Aliments contenant 1 mg d’acide phytanique
- - Riz blanc bouilli 100 g
- - Spaghettis cuits 85 g
- - Pain 60 g
- - Corn Flakes 85 g
- - Rice Krispies 35 g
- - Sugar Puffs 95 g
- - Weetabix 65 g
- - Crème au lait écrémé 120 g
- - Pois chiches en boîte 50 g
- - Pommes de terre
- Bouillies 150 g
- Chips 100 g
- Rôties ou sautées 120 g
Aliments contenant 2 mg d’acide phytanique
- Fromage blanc à la louche 100 g
- Veau mijoté 90 g
- Porc mijoté 50 g
- Jambon blanc 40 g
- Morue grillée 50 g
- Haddock fumé 80 g
- Crabe 40 g
- Haricots blancs en boîte 70 g
- Pois chiches en boîte 100 g
Liste de poissons maigres (taux de lipides < 2 %) : brochet, carpe, goujon, tanche, bar, colin, cabillaud, carrelet, dorade, églefin, limande, sole, merlan, raie ; coquillages (coques, moules) ; seiche ; crabe ; crevettes ; grenouille.
Conseil génétique
Un patient atteint de maladie de Refsum n’a aucune chance de transmettre la maladie à ses enfants, sauf si l’autre parent a un lien de parenté (consanguinité).
Les parents d’enfants atteints de maladie de Refsum ont 25% de risque d’avoir d’autre enfants atteints de la même maladie.
Un diagnostic pré-natal peut être proposé dans les familles à risque.
Bibliographie
Le site web du péroxysome (en anglais)
Jansen GA, Wanders RJ, Watkins PA, Mihalik SJ. Phytanoyl-coenzyme A hydroxylase deficiency -- the enzyme defect in Refsum's diseas N Engl J Med 1997 Jul 10;337(2):133-4
Jansen GA, Ofman R, Ferdinandusse S, Ijlst L, Muijsers AO, Skjeldal OH, Stokke O, Jakobs C, Besley GT, Wraith JE, Wanders RJ. Refsum disease is caused by mutations in the phytanoyl-CoA hydroxylase gene Nat Genet 1997 Oct;17(2):190-3
Masters-Thomas A, Bailes J, Billimoria JD, Clemens ME, Gibberd FB, Page NG. Heredopathia atactica polyneuritiformis (Refsum's disease): 1. Clinical features and dietary management J Hum Nutr 1980 Aug;34(4):245-50
Mihalik SJ, Morrell JC, Kim D, Sacksteder KA, Watkins PA, Gould SJ. Identification of PAHX, a Refsum disease gene Nat Genet 1997 Oct;17(2):185-9.
Mukherji M, Chien W, Kershaw NJ, Clifton IJ, Schofield CJ, Wierzbicki AS, Lloyd MD. Structure-function analysis of phytanoyl-CoA 2-hydroxylase mutations causing Refsum's disease. Hum Mol Genet. 2001 Sep 1;10(18):1971-82.
Refsum, S.; Salomonsen, L.; Skatvedt, M. : Heredopathia atactica polyneuritiformis in children. J. Pediat. 35: 335-343, 1949.
Refsum S. Heredopathia atactica polyneuritiformis phytanic-acid storage disease, Refsum's disease:" a biochemically well-defined disease with a specific dietary treatment" Arch Neurol 1981 Oct;38(10):605-6
Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular basis of inherited disease, 7th edition. New York: McGraw-hill, Inc.; 1995. 2351-2369.
Traboulsi E. Genetic diseases of the eye Oxford 1998.
Wanders RJ, Jansen GA, Skjeldal OH. Refsum disease, peroxisomes and phytanic acid oxidation: a review J Neuropathol Exp Neurol 2001 Nov;60(11):1021-31.
Wierzbicki AS, Lloyd MD, Schofield CJ, Feher MD, Gibberd FB. Refsum's disease: a peroxisomal disorder affecting phytanic acid alpha-oxidation J Neurochem 2002 Mar;80(5):727-35.

Sigvald Refsum
By courtesy of Refsum family
Je remercie la famille de la jeune fille porteuse de cette maladie, qui m'a sensibilisé à cette pathologie, ce qui m'a incité à écrire ces quelques lignes.
jmm

![]()
Suivez nous sur...
![]()
SNOF
10 rue Schweighaeuser
CS 40028
67080 STRASBOURG Cedex
Tél. 03 88 35 01 09
Fax. 03 88 25 51 90